神経変性疾患におけるTNF-αとアストロサイト
アストロサイトにおけるTNF-αシグナル伝達の概要、神経変性におけるその役割、およびこの経路を標的とする治療戦略について。
TNF-αシグナル伝達は、神経変性疾患においてアストロサイト機能にどのような影響を及ぼすのでしょうか?
アストロサイトは腫瘍壊死因子-α(TNF-α)に対し、TNFR1とTNFR2という二つの受容体システムを介して応答します。これらの受容体の相反する作用が、神経毒性と修復のバランスを確立しています。アストロサイトにおけるTNFR1の活性化は、実験的自己免疫性脳脊髄炎(EAE)モデルで示されるように、生体内においてシナプス機能障害や記憶障害を誘導するのに十分です。このモデルでは、海馬の障害がグリア細胞のTNFシグナル伝達と直接関連しています(Habbas, 2015 )。慢性予測不能軽度ストレス(CUMS)マウスなどの前臨床慢性ストレスモデルでは、TNFR1の上昇が星状膠細胞増殖と神経細胞アポトーシスを特徴とするうつ病様症状と関連しており、薬理学的または遺伝学的にTNFR1を遮断することでこれらの影響が逆転します(Gao, 2024 )。これに対し、アストロサイトにおけるTNFR2シグナル伝達は、脱髄状態において炎症促進プログラムを抑制することで反応性アストロサイト増殖を制限し、再髄鞘形成を促進します。同時に、生理的状態下では海馬のシナプス機能、可塑性、認知機能を支持します(Raphael, 2019 ;Carney, 2025 )。
この受容体の機能分化は治療的機会を生み出します:
- 主にTNFR1を活性化する可溶性TNF(sTNF)を選択的に中和することで、脱髄モデル(例: クプリゾン脱髄モデル)における再髄鞘形成が促進され、同時に膜貫通型TNF(tmTNF)-TNFR2経路を介した有益なシグナル伝達は維持されます(Karamita, 2017 )。
- この分岐を利用しつつ非選択的TNF遮断による合併症を回避するため、選択的TNFR2アゴニストが活発に開発中です(Pegoretti, 2023 )。
これらの知見は、アストロサイトにおける分子スイッチとしてのTNFR1とTNFR2の役割を浮き彫りにし、神経毒性と修復プログラムのバランスを左右する存在であることを示しています。さらに、受容体選択的標的化が有望な治療アプローチであることを強調しています。
受容体特異的効果を超えて、TNF-αはアストロサイトの状態遷移を調整し、グリア環境を保護プログラムと神経毒性プログラムの間で移行させます。顕著な例として、IL-1α、TNF-α、C1qからなるサイトカイン三連星が挙げられ、これらは恒常性アストロサイトを補体C3発現を特徴とする神経毒性A1状態へと移行させます。この経路の阻害はA1状態への転換を防ぎ、ALSモデルにおける生存期間を延長させ、アストロサイトにおけるTNFシグナル伝達が疾患進行に直接関与することを示しています(Liddelow, 2017 ;Guttenplan, 2020 )。しかしながら、この表現型が常に有害であるとは限りません。プリオン病においては、C3陽性アストロサイトの除去が変性を悪化させ、疾患や状況に応じた特異的な役割を強調しています(Hartmann, 2019 )。 ヒトiPS細胞由来アストロサイトにおいても同様の動態が再現され、TNF-αは単独またはIL-1βとの共刺激によりNF-κB活性化とC3発現亢進を引き起こし、グルタミン酸クリアランス機能を障害します(Hyvärinen, 2019 )。最近のマルチオミクス解析により、ヒトのアルツハイマー病(AD)および筋萎縮性側索硬化症(ALS)の脳組織において、A1様アストロサイトが神経細胞喪失部位周辺に集積することが示され、その病原性ポテンシャルが裏付けられました(Escartin, 2021 )。パーキンソン病(PD)においては、TNF-αに曝露されたアストロサイトとα-シヌクレイン線維に曝露されたアストロサイトは異なる反応パターンを示しますが、いずれもミトコンドリア機能障害という共通の終点に収束し、これが疾患進行に寄与しています(Russ, 2021 )。これらの知見を総合すると、TNF-αはアストロサイトの状態遷移を促進する主要な要因であり、状況依存的な方法で疾患に影響を与える神経毒性プログラムへと導くことが示されています。
ミクログリアにおけるTNF-αの機能および神経変性疾患への寄与に関する総説については、以下をご参照ください:TNF-αと神経変性疾患におけるミクログリア
アストロサイトの表現型形成を超えて、TNF-αは神経細胞および血管の健康に不可欠な主要なアストロサイト機能を直接的に変化させます:
- TNF-αは、TNFR1/NF-κB経路を介してEAAT2/GLT-1の発現を抑制することでグルタミン酸取り込みを減少させ、運動ニューロンへの興奮毒性ストレスを増大させます(Jiang, 2019 )。
- ヒトiPS細胞由来アストロサイトにおいて、TNF-αおよびその他の炎症性サイトカインはグルタミン酸クリアランスを低下させ、免疫反応性シグネチャを増強します(Hyvärinen, 2019 )。
- TNF-αはグリア伝達を促進します。小脳回路において、ベルグマン膠細胞におけるTNFR1の活性化は、mGluR1依存性メカニズムを介してグルタミン酸放出を増加させ、神経細胞の興奮性を変化させます(Shim, 2018 )。
- 炎症状態におけるTNF-STAT3軸は血液脳関門(BBB)の完全性を損ない、炎症が誘導されたパーキンソン病アストロサイトは微小血管形態形成をサポートできません。この欠陥はMEK1/2阻害により逆転します(de Rus Jacquet, 2023 )。
これらの知見は、TNF-αが炎症状態の変化を促進するだけでなく、グルタミン酸の恒常性維持、神経細胞とグリア細胞のコミュニケーション、血管の安定性といったアストロサイトの重要な役割を損ない、それによって神経変性プロセスを加速させることを示しています。
これらの多様な結果は、TNF受容体の下流にあるシグナル伝達ネットワークによって説明されます。このネットワークは、炎症および代謝のハブをアストロサイトの状態制御に結びつけています:
- NF-κB活性化(RelA/p65)は炎症性転写プログラムを駆動し、ミトコンドリア機能障害に寄与します。これらの生体エネルギー障害はヒト星状細胞モデルで確認されています(Russ, 2021 )。
- MAPKシグナル伝達モジュールはさらにTNF経路と交差します:ERK/MAPK活性はパーキンソン病アストロサイトにおける血液脳関門(BBB)の支持機能を調節します。
- ミクログリアTNF下流のJNK活性化は、アストロサイトにおけるCXCL1産生を誘導し、生体内におけるニューロン-グリア間コミュニケーションを再構築します(Zhang, 2022 ;de Rus Jacquet, 2023 )。
以上を総合すると、NF-κBおよびMAPKハブと統合されたTNFR1とTNFR2の二分性は、TNF-αをアストロサイト状態遷移のマスターレギュレーターとして位置づけ、神経変性疾患全体において神経毒性プログラムまたは修復プログラムのいずれかを調整していると考えられます(Raphael, 2019 )。
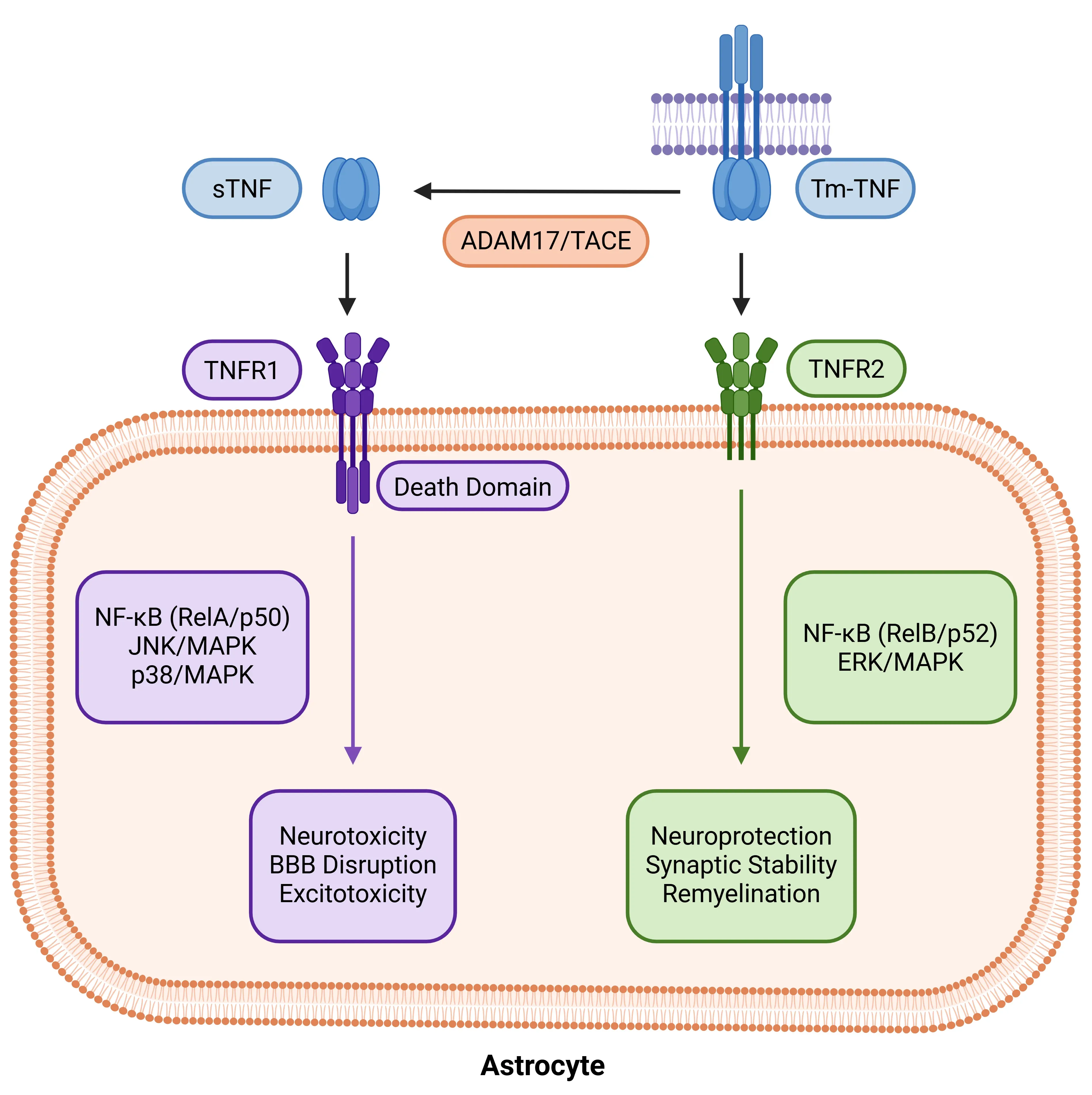
アストロサイトにおけるTNF-α受容体の二分性。アストロサイトが TNF-αに反応する仕組みは、リガンドの形態と受容体の特異性に依存します。ADAM17/TACEによってtmTNFから切断されたsTNFはTNFR1を活性化し、NF-κB(RelA/p50)、JNK、p38 MAPK経路を誘発します。これらは神経毒性と血液脳関門(BBB)の破壊を促進します。一方、tmTNFは主にTNFR2に結合し、非典型的なNF-κB(RelB/p52)およびERK/MAPKを介したシグナル伝達により、神経保護、シナプス安定化、再髄鞘形成を促進します。この二分性は、TNF-αを損傷と修復の間でアストロサイトの状態を調節する重要な因子として位置づけています。
反応性アストロサイトは、TNF-αを介した神経炎症においてどのような役割を果たすのでしょうか?
TNF-α下における反応性アストロサイトは、単純なグリア活性化を超えた複数の有害な役割を担います。ここでは、これらの状態が様々な疾患環境において、神経炎症、血管機能障害、および直接的な神経細胞損傷にどのように寄与するかを説明します。
グリア毒性と血管機能障害
星状細胞の反応性は、炎症を起こした脳内におけるミクログリアと星状細胞のコミュニケーションの重要な増幅因子として機能するTNF-αによって大きく形作られます。ミクログリア由来のTNF-αは、IL-1αおよびC1qと相まって、in vitroおよびin vivoの両方で、恒常性アストロサイトから神経毒性のあるA1状態への転換を十分に引き起こし、TNF-αをアストログリアーシスの主要な誘導因子として確立しています。このIL-1α/TNF/C1qの三要素を中和することでA1状態への転換を防ぎ、神経細胞およびオリゴデンドロサイト(少突膠細胞)の生存を保持することが確認されており、TNF-α依存性の反応性が単なる疾患の副産物ではなく病態の原因であることを裏付けています(Liddelow, 2017 )。海馬回路において、TNF-αによる星状細胞のTNFR1活性化は、興奮性シナプスを再構築し文脈記憶を損なう星状細胞-神経細胞カスケードを開始します(Habbas, 2015 )。
血管界面において、TNFは星状細胞をSTAT3依存性の炎症反応状態に誘導し、血液脳関門(BBB)の完全性を損ない、血管内皮炎症を誘発する因子を放出させます(Kim, 2022 )。この血管障害の証拠に基づき、最近のヒト脳チップ研究では、TNFによって活性化されたアストロサイトがバリア機能を不安定化させるだけでなく、微小血管の形態形成を損なうことも示されています。この欠陥はMEK1/2阻害により可逆的です(de Rus Jacquet, 2023 )。
以上を総合すると、TNF-α反応性アストロサイトは、毒性のあるグリア状態を助長し、シナプス伝達を阻害し、血液脳関門および微小血管の完全性を弱体化させることで、神経炎症性損傷を増幅させる主要な要因として機能していることがわかります。
神経炎症の炎症性および酸化ストレス性駆動因子
アストロサイトがTNF-α駆動型の反応状態を採用すると、グリア細胞領域をはるかに超えて損傷を拡散させる炎症および酸化ストレスの活性源となります。中心的なメカニズムはNF-κB経路の活性化であり、これにより以下の炎症促進性メディエーターの産生が誘導されます:
これらのシグナルは白血球を動員し、中枢神経系(CNS)の局所免疫活性を増幅させます(Giovannoni, 2020 )。
並行して、反応性アストロサイトは活性酸素種(ROS)および活性窒素種(NOS)を産生し、TNF-αシグナル伝達を酸化ストレス経路に結びつけ、神経細胞および血管の恒常性をさらに不安定化させます(Ding, 2021 )。ミクログリアとの双方向的な相互作用により、これらの反応はさらに強まります。アストロサイトにおけるNF-κBの活性化は、ミクログリアの増殖と白血球の浸潤を促進するのに十分であり、アストロサイトが受動的に反応するのではなく、能動的に神経炎症をエスカレートさせていることを示しています(Ouali Alami, 2018 )。
参照:ミクログリアおよびアストロサイトにおけるミトコンドリア機能障害
ヒトiPS細胞由来アストロサイトモデルはこれらの特徴を再現しており、IL-1βとTNF-αの併用刺激によりNF-κBの核移行、形態学的リモデリング、および免疫反応性表現型の獲得が誘導されます(Hyvärinen, 2019 )。これらの実験モデルと一致して、ヒトのアルツハイマー病(AD)および多発性硬化症(MS)組織からの単一核RNA-seq(snRNA-seq)は、NF-κB駆動型アストロサイト状態がプラークや病変の周囲に集積していることを示しており、その直接的な臨床的関連性を強調しています(Escartin, 2021 )。
これらの知見を総合すると、TNF-α反応性アストロサイトは強力な炎症および酸化ストレスのエンジンとして機能し、神経炎症カスケードを積極的に増幅させ、神経細胞および血管の脆弱性を悪化させることが示されています。
神経毒性と回路機能障害
TNF-αによって駆動されるアストロサイトの反応性は、その正常な支持的役割を奪うだけでなく、神経細胞およびオリゴデンドログリア細胞損傷の積極的な発生源ともなります。機能的には、A1アストロサイトは正常なシナプス形成促進サポートを失い、神経細胞およびオリゴデンドロサイト死を誘発する因子を分泌します。これにより、TNF-α誘導性反応状態と神経変性との直接的な関連性が示されています(Liddelow, 2017 )。一つのメカニズムとして、APOE/APOJリポ粒子による飽和脂質の分泌が挙げられます。脂質代謝酵素ELOVL1を阻害することでこの毒性は防止され、アストロサイトの脂質代謝がTNF誘導性神経細胞死に関与していることが示唆されています(Guttenplan, 2021 )。
回路レベルでは、単独またはIL-6と組み合わせて持続的にTNF-αに曝露すると、ヒト神経細胞-星状細胞共培養系において神経細胞の発火パターンが乱れ、炎症時の認知機能障害や行動障害の根底にある可能性のある異常なリズムが生じます(Goshi, 2025 )。同様の障害は生体内でも観察されており、慢性的なTNF-α曝露は海馬の過興奮性とてんかん様活動を促進します(Vezzani, 2020 )。
全体として、TNF-αに反応するアストロサイトは、神経細胞の喪失とネットワークの不安定化の重要な要因として浮上しており、グリア細胞の反応性と神経変性疾患の進行を直接結びつけています。
これらの証拠は、TNF-αがアストロサイトの生物学的特性を根本的に再構築し、中枢神経系の恒常性維持の守護者から機能不全の中心的な原動力へと変容させることを示しています。炎症、血管障害、神経細胞の脆弱性の交差点にアストロサイトを位置づけることで、TNF-αはアストロ細胞を受動的な応答者ではなく、神経変性進行の決定的な調節因子として明らかにします。この視点は、アストロ細胞を背景的な存在から、慢性神経炎症との闘いにおける重要な治療標的へと再定義するものです。
TNF-αは、アストロサイトを介して神経変性疾患の進行にどのように関与するのでしょうか?
慢性的なTNF-α曝露は、アストロサイトを徐々に有害な状態へと追い込み、その遺伝子発現、構造、代謝を変化させます。低濃度であっても、持続的なTNF-αシグナル伝達は星状細胞の肥大化、炎症性遺伝子の再配線、代謝ストレスを引き起こします。これにより、研究者は星状細胞の反応性を単に「保護的または毒性」と捉えるのではなく、変化する状態のスペクトルとして見るようになりました(Escartin, 2021 )。
TNF-αを含む炎症性ミクログリアシグナルは、神経毒性を持つA1様アストロサイトへの移行を促進し、神経変性疾患における慢性的な自然免疫活性化とアストロサイト機能障害との直接的な関連性を示しています(Liddelow, 2017 )。ヒトiPS細胞由来アストロサイトにおいても同様の過程が再現され、TNF-α(±IL-1β)がNF-κBを迅速に活性化し、形態および炎症性遺伝子ネットワークを再構築します(Hyvärinen, 2019 )。パーキンソン病に関連するヒトアストロサイトにおいて、TNF-αまたはα-シヌクレイン線維は、ミトコンドリア呼吸障害を特徴とする重畳した免疫代謝状態へアストロサイトを誘導し、サイトカインへの長期曝露が細胞のエネルギーバランスを損なうことを示しています(Russ, 2021 )。
これらの知見を総合すると、慢性的なTNF-αシグナル伝達はアストロサイトに持続的な炎症および代謝負荷を刻印し、その支持的役割を弱め、神経変性疾患の進行に積極的に寄与する機能不全状態へと移行させることが示されています。
シナプスレベルでは、TNF-αはアストロサイトのグルタミン酸処理とグリア伝達を変化させ、神経回路を不安定化し興奮毒性に対する脆弱性を高めます:
- アストロサイトにおけるTNFR1の活性化のみでも、自己免疫性脱髄疾患において興奮性伝達を変化させ海馬記憶を障害することが示されており、ネットワーク機能不全におけるアストロサイトTNF-αの直接的な因果的役割が実証されています(Habbas, 2015 )。
- TNF-αは、TNFR1-NF-κB経路を介して、EAAT2/GLT-1輸送体の発現を減少させ、グルタミン酸取り込みを弱め、運動ニューロンを毒性のある過剰刺激に晒します(Jiang, 2019 )。
- ヒトiPS細胞由来アストロサイトは、TNF-αおよびIL-1βによる共刺激により、グルタミン酸クリアランスの障害とより強い炎症活性化を示します(Hyvärinen, 2019 )。
- TNF-αは、ベルグマングリアのグルタミン酸放出を増加させることで小脳回路におけるグリア伝達を増強し、mGluR1シグナル伝達を介してプルキンエ細胞の興奮性を高め、発火パターンの不安定化を引き起こします(Shim, 2018 )。
- 持続的なTNF-α(±IL-6)曝露は、マイクロ電極アレイ(MEA)上の人工神経細胞-アストロサイト共培養系において神経細胞の放電ダイナミクスを乱し、慢性的なサイトカインが新たな回路不整脈を引き起こすことを示唆しています(Goshi, 2025 )。
- 生体内研究では、TNF-αの過剰発現が発作感受性を高め、認知機能低下を加速させることが示されており、星状細胞の機能不全が広範なネットワーク不安定性に関連していることが明らかになりました(Vezzani, 2020 )。
これらの知見を総合すると、TNF-αによる星状細胞の機能障害がグルタミン酸の調節とシナプスバランスを損ない、広範な回路不安定性と進行性の認知障害を引き起こすことが明らかになりました。
神経血管単位(NVU)内では、TNF-αによって活性化されたアストロサイトが炎症シグナルを伝達し、血液脳関門(BBB)の完全性を弱め、血管機能を損ないます。ヒトおよびex vivoモデルにおいて、TNF-αはアストロサイトをSTAT3依存性の反応状態へと誘導し、SERPINA3(α1-アンチキモトリプシン、急性期セリンプロテアーゼ阻害因子としても知られる)の発現亢進を特徴とします。これにより血液脳関門の完全性が低下し、内皮機能障害が促進されます(Kim, 2022 )。
パーキンソン病関連LRRK2変異を有するアストロサイトは、ヒト脳チップBBBシステムにおいて炎症促進プロファイルを示し、微小血管形態形成の維持に失敗します。この欠損はMEK1/2阻害により回復します(de Rus Jacquet, 2023 )。これらの知見は、星状細胞の終足およびその分泌分子がBBBの安定性を調節し、これらの界面における炎症性リモデリングが神経変性疾患における血管機能障害に寄与するという広範な証拠と一致しております(Yue, 2023 )。これらのメカニズムに関する知見と一致して、ヒト多発性硬化症病変における空間トランスクリプトミクス解析は、TNF刺激を受けた星状細胞が血管周囲領域に集積し、サイトカイン豊富なニッチを形成することでBBB構造を積極的に不安定化させることを示しています(Mazziotti, 2024 )。
これらの知見は、TNF-αによって誘導されたアストロサイトがBBBの完全性と血管の健康を積極的に破壊し、神経変性疾患における神経血管機能障害の主要な要因となることを示しています。
主要な神経変性疾患において、TNF-αによって駆動されるアストロサイトプログラムは、疾患進行の共通した増幅因子として作用します。炎症性シグナル伝達をシナプス機能障害、代謝異常、血管機能障害と結びつけることで、アストロサイトはTNF-αシグナルを局所的なメカニズムに変換し、神経細胞の脆弱性と組織の衰退を加速させます。これらの核心的なプロセスは疾患間で共通していますが、TNF-αに反応するアストロサイトが疾患特異的な環境と相互作用する方法は大きく異なります。以下のセクションでは、アストロサイトを介したTNF-αシグナル伝達が、アルツハイマー病、パーキンソン病、ALS、および多発性硬化症(MS)の進行にどのように寄与するかを説明します。
アルツハイマー病(AD)
TNF-αは、アミロイドβ病変、神経炎症、シナプス機能障害を結びつける、ADにおける重要なメディエーターとして注目されています。軽度認知障害(MCI)およびAD患者の脳脊髄液(CSF)と血清の両方において、TNF-αレベルの上昇が一貫して報告されており、より高いベースライン濃度は認知機能低下の加速および認知症への進行リスクの増加を予測します(Kim, 2017 ;Lista, 2024 )。重要なことに、これらの生化学的変化は星状細胞の変化と並行して起こります。反応性アストロサイトバイオマーカー(CSF GFAPやYKL-40など)は、アミロイドβおよびタウが海馬萎縮と認知機能に及ぼす影響を媒介し、生体内のヒトにおいてアストロサイトの反応性と下流の神経変性を結びつけています(Ferrari-Souza, 2022 ;Pelkmans, 2024 )。神経病理学的研究により、反応性アストロサイトがβアミロイド斑および神経原線維変化の周囲に集積し、斑の核心部に侵入して病変の封じ込めと拡散の両方に寄与していることがさらに明らかになっています(Perez-Nievas, 2018 )。
アミロイドβプラーク微小環境におけるアストロサイト形態の分析については、以下をご参照ください:アストロサイトとアミロイドβ アルツハイマー病マウスモデル
TNF-αがアルツハイマー病に寄与する中心的なメカニズムには、アストロサイトにおけるアミロイド生成プロセシングを促進する能力が関わっています。具体的には、TNF-αシグナル伝達は:
- アミロイド前駆体タンパク質(APP)およびBACE1の発現を亢進させ、βアミロイド生成とプラーク蓄積を促進します(Zhao, 2011 )。
- 興奮性-抑制性のバランスを変化させることでシナプス恒常性を破壊します:
- グルタミン酸作動性伝達を増強します
- GABA作動性抑制を減少させます
- AMPA受容体の細胞表面への輸送を増加させ、
これらの作用が相まって、海馬回路を興奮毒性に脆弱にします(Pribiag, 2013 ;Heir, 2020 )。この興奮毒性損傷は、アストロサイトを介したグルタミン酸放出によってさらに増幅され、神経細胞の生存を損ないます(Santello, 2011 )。
アストロサイト自体は受動的な応答者ではなく、アルツハイマー病(AD)におけるTNF-αシグナル伝達の主要な駆動因子です。実験的証拠は以下の通りです:
- ミクログリアを欠く海馬培養系ではTNF-αシグナル伝達が持続しますが、星状細胞特異的TNF欠損により消失します。これにより、活動依存性TNF放出に星状細胞が不可欠であることが確立されました(Heir, 2024 )。
- 星状細胞のNF-κB活性化は、グルタミン酸の流出に対するTNF産生を調節し、サイトカイン駆動型回路リモデリングの中心に星状細胞を位置づけています。
- 星状細胞のNF-κBシグナル伝達を調節することで、アルツハイマー病モデルにおける神経細胞およびシナプスの結果が変化します(Jong Huat, 2024 )。
- アストロサイトにおけるグルタミン酸処理機能の障害、特にEAAT2/GLT-1トランスポーター機能の低下は、TNFによる興奮毒性と相まって神経変性を悪化させます(Wood, 2022 )。
遺伝学的研究はTNF-αの病原性役割を裏付けています。TNF-αプロモーター領域のG-308A多型は転写活性とタンパク質発現を増加させ、複数のメタ解析でアルツハイマー病感受性の増大と関連付けられています(Wang, 2015 )。この変異体はAPOE-ε4遺伝子型と相乗的に作用し、炎症性プライミングと疾患進行を加速させる可能性があります(Contreras, 2020 )。これは、散発性および家族性ADに共通する要素としてTNFシグナル伝達の重要性を強調するものです。
治療研究は、TNF-αを標的とする治療法の応用可能性を強調しています。データは以下の通りです:
- エタネルセプトやアダリムマブなどのTNF阻害薬で治療を受けた全身性炎症性疾患患者では、未治療群と比較して認知症発症率が低下しています。
- 脊髄周囲エタネルセプト投与を用いた小規模パイロット試験および症例報告では、AD患者において迅速かつ持続的な認知機能改善が報告されています。
- 皮下投与エタネルセプトを用いた大規模ランダム化比較試験では、大きなバイオ製剤の血液脳関門(BBB)透過性の低さが原因と考えられ、有意な効果は確認されませんでした。
- 血液脳関門透過性TNF阻害剤を用いた前臨床研究では、ADマウスモデルにおいてβアミロイドおよびタウ病理の減少と認知機能改善が確認され、TNF標的療法の根拠が強化されています(Plantone, 2024 )。
これらの知見を総合すると、TNF-αはアストロサイトを介したメカニズムを通じて、ADにおけるβアミロイド蓄積、興奮毒性、神経変性の中心的な駆動因子であり、生物学的に妥当な治療標的であることが示されています。臨床データ、特に血液脳関門(BBB)送達に関するものは依然として予備的段階ですが、TNF-α阻害は疾患修飾療法として有望な道筋を示しています。
パーキンソン病(PD)
アストロサイト由来のTNF-αはパーキンソン病(PD)において中心的な役割を果たし、α-シヌクレイン病理と神経炎症増幅および神経細胞脆弱性を結びつける媒介因子として浮上しています。パーキンソン病患者では、脳、脳脊髄液、血液中においてTNF-αおよび可溶性受容体が一貫して上昇しており、これはドーパミン作動性神経変性、認知機能低下、および疾患の重症度全体と相関しています(Liu, 2022 )。 アストロサイト内のα-シヌクレインの蓄積は 、ミトコンドリアおよび小胞体の機能を損なうだけでなく、反応性の炎症促進型表現型を促進します(Wang, 2021 )。重要なことに、α-シヌクレイン封入体を含む星状細胞は高レベルのTNF-αを分泌し、このサイトカインをタンパク質病変と炎症増幅を結びつける中心的なエフェクターとして位置づけています(Lee, 2010 )。
星状細胞由来のTNF-αは、パーキンソン病(PD)におけるグリア細胞の反応性のフィードフォワードループに寄与します。
パーキンソン病およびその他のシヌクレイン病において、α-シヌクレインがミクログリアおよびアストロサイトに与える影響に関する詳細なレビューについては、以下をご参照ください:パーキンソン病におけるミクログリア、アストロサイト、およびα-シヌクレイン
IL-1βおよびIL-6と並行して、星状細胞由来のTNF-αは:
- ミクログリアの活性化を増幅し、それによってドーパミン作動性ニューロンの喪失を加速させます。
- 神経細胞の脆弱性を悪化させます。
- アストロサイト主導のサイトカインシグナル伝達と進行性黒質線条体変性を結びつけるグリア反応性のフィードフォワードループを形成します(Wang, 2023 )。
実験的パーキンソン病モデルでは、明らかな神経細胞喪失以前に、IFN-γとTNF-αの両方がミクログリアおよびアストロサイト活性化を持続させるために必要であることが示されており、TNF-αがグリア細胞主導の神経炎症を誘発する役割を強調しています(Barcia, 2011 )。さらに、機構的研究により、調節因子RGS5が以下の作用を有することが明らかになりました:
- アストロサイトにおいてTNFR2シグナル伝達を保護的から炎症促進的へ転換させることを明らかにしました。
- TNFR1の活性化を増幅します。
- パーキンソン病モデルにおいてα-シヌクレイン凝集、神経変性、および死亡率を促進します(Yin, 2023 )。
アストロサイトが神経毒性を持つA1状態へ転換する過程も、TNF-αが不可欠な病態メカニズムの一つです。ミクログリアが放出するIL-1α、TNF-α、C1qが共同でアストロサイトをこの有害な表現型へ転換させます。A1アストロサイトはPD患者の死後脳で確認されています(Liddelow, 2017 )。パーキンソン病動物モデル(散発性PDのα-syn PFFモデルおよびhA53Tトランスジェニックマウスモデル)において、病理学的α-シヌクレイン( )もA1アストロサイトの形成を促進し、この過程を阻害することでドーパミン作動性ニューロンが保護され運動機能が維持されます(Yun, 2018 )。これらのA1アストロサイトは:
- 栄養的・シナプス的サポートを提供する能力を失います。
- 補体成分や活性酸素種(ROS)を含む神経毒性因子を分泌します。
- 神経細胞損傷を悪化させます(Liddelow, 2017 )。
ヒト研究により、アストロサイト由来TNF-αのパーキンソン病における病原性役割が確認されています。ヒトパーキンソン病中脳の単一核RNA-seq解析では、CD44高発現アストロサイト表現型とサイトカインシグナル経路を伴う汎グリア活性化が明らかとなり、黒質ニッチにおける慢性炎症状態と一致しています(Smajić, 2022 )。培養ヒト星状細胞において、TNF-αおよびα-シヌクレイン線維は、ミトコンドリア呼吸障害を伴う免疫反応性状態を引き起こします。これは、PDの病態形成と一致するTNF応答性代謝障害を浮き彫りにしています(Russ, 2021 )。この病原性カスケードは、補体C4によってさらに増幅され、以下の作用を示します:
- α-シヌクレインに対する星状細胞の炎症促進応答を増強します。
- 神経細胞のアポトーシスとシナプス病変を促進します(Zou, 2025 )。
さらに、炎症状態に誘導されたヒト星状細胞は、脳チップモデルにおいて微小血管の形態形成を阻害し、血液脳関門(BBB)の完全性を損なうことが示されています(de Rus Jacquet, 2023)。そのメカニズムとして、TNFは星状細胞をSTAT3依存性の急性期様状態へと誘導し、全身性炎症を脳内皮細胞へ伝達することで、パーキンソン病におけるBBB破壊を促進します(Kim, 2022 )。
これらの知見を総合すると、アストロサイト由来TNF-αは、α-シヌクレイン病理、グリア増幅、シナプス機能障害、およびPDにおける血管障害を結びつける中心的なメカニズムとして位置づけられます。
筋萎縮性側索硬化症(ALS)
星状細胞由来TNF-αシグナル伝達は、ALSにおける運動ニューロンの脆弱性を引き起こす早期かつ持続的な要因として明らかになっております。ALS患者の脳脊髄液(CSF)および血液中では、TNF-αのレベルが持続的に上昇しており、これは疾患の進行と相関しています(Jiang, 2022 )。同様に、SOD1G93Aマウスにおいても、症状発現前から脊髄においてTNF-αおよびその受容体が上調節されており、この経路が早期病態形成に寄与していることが示唆されています(Brambilla, 2016 )。これらのモデル由来の星状細胞-運動ニューロン共培養系では、運動ニューロンにおける膜結合型TNF-αが増加する一方でTNFR2レベルが低下し、受容体シグナル伝達の不均衡が生じます。特に注目すべきは:
- TNFR2(TNFR1ではない)を欠損させると運動ニューロンの生存が回復し、mTNF-α-TNFR2軸が神経毒性の主要な媒介因子であることを明らかにしました(Tortarolo, 2015 )。
- 一方、TNFR1シグナル伝達は、アストロサイト由来神経栄養因子(GDNF)の放出を介した可能性のある保護効果を発揮するかもしれません。なぜなら、その欠損は予後を悪化させるからです(Brambilla, 2016 )。
これらの知見は、TNFR1およびTNFR2がALS病態に寄与する程度が、単離共培養系と複雑な 生体内環境とでは大きく異なり、状況に強く依存することを強調しています。さらに、TNFR1とTNFR2はいずれもアポトーシスシグナル調節キナーゼ1(ASK1)/p38MAPK経路を活性化し、これは運動ニューロン死に決定的に関与しています。p38 MAPKを阻害すると、SOD1G93Aマウス由来星状細胞/運動ニューロン共培養モデルにおいて運動ニューロンが保護され、この経路がALSにおけるTNFシグナル伝達の主要な下流エフェクターであることを示しています(Dewil, 2007 )。並行して:
- 変異型FUSを有する星状細胞において、TNF-αはNF-κBを活性化し、GluA2の組み込みを減少させることでAMPA受容体サブユニットの構成を変化させます。
- この変化により、運動ニューロンにおけるカルシウム透過性と興奮毒性ストレスが増大します( )(Kia, 2018 )。
- FUS-ALSモデルにおける遺伝的TNF-α欠損または薬理学的阻害は運動行動と神経細胞生存を回復させ、アストロサイト由来TNF-αが直接的な毒性シグナルであることを明らかにしました(Jensen, 2022 )。
アストロサイト由来のTNF-αは炎症環境を形成し、ALS病態を悪化させるグリア細胞および免疫応答を促進します。主なメカニズムは以下の通りです:
- アストロサイトにおける誘導性NF-κBの活性化は、ミクログリアの増殖と末梢白血球の動員を促進し、症状の進行を加速させます(Ouali Alami, 2018 )。
- ミクログリアのIL-1α/TNF/C1qシグナル伝達三連鎖は神経毒性A1型アストロサイト表現型を誘導し、この軸を阻害することでSOD1G93Aマウスの生存期間が延長されます(Guttenplan, 2020 )。
- SOD1変異を有するアストロサイトは、ミトコンドリアストレス、プロテアソーム機能障害、および自然免疫活性化によって駆動される反応性で機能不全な状態をとり、この状態はTNF-αシグナル伝達によってさらに悪化します。
- 線維芽細胞成長因子4(FGF4)は星状細胞の恒常性を一時的に回復させることができますが、TNF-αはNF-κB活性を持続させることでこの保護作用を無効化し、運動ニューロンを脆弱な状態に置きます(Velasquez, 2024 )。
星状細胞由来のTNF-αはグルタミン酸の恒常性を乱し、ALSにおける興奮毒性ストレスをさらに増強します。具体的には、星状細胞のTNFR1を介したTNF-αシグナル伝達は:
- EAAT2/GLT-1の発現を抑制します。
- グルタミン酸放出を促進します。
- グルタミン酸のクリアランス障害と運動ニューロンへの興奮毒性ストレスを引き起こします。このメカニズムはヒトのALS病理と一致しています(Jiang, 2019 )。
この障害は、TNFによるAMPA受容体構成の変化と相乗的に作用し、カルシウム依存性興奮毒性を増強します(Kia, 2018 ;Jensen, 2022 )。
総括しますと、アストロサイト由来のTNF-αは、ALSにおける炎症、受容体バランスの乱れ、興奮毒性の三者を結ぶ収束点として機能します。炎症カスケードの増幅、グルタミン酸処理の変容、興奮性受容体の透過性増加を同時に引き起こすことで、TNF-αは運動ニューロンの変性を加速し、疾患進行を促進するのです。
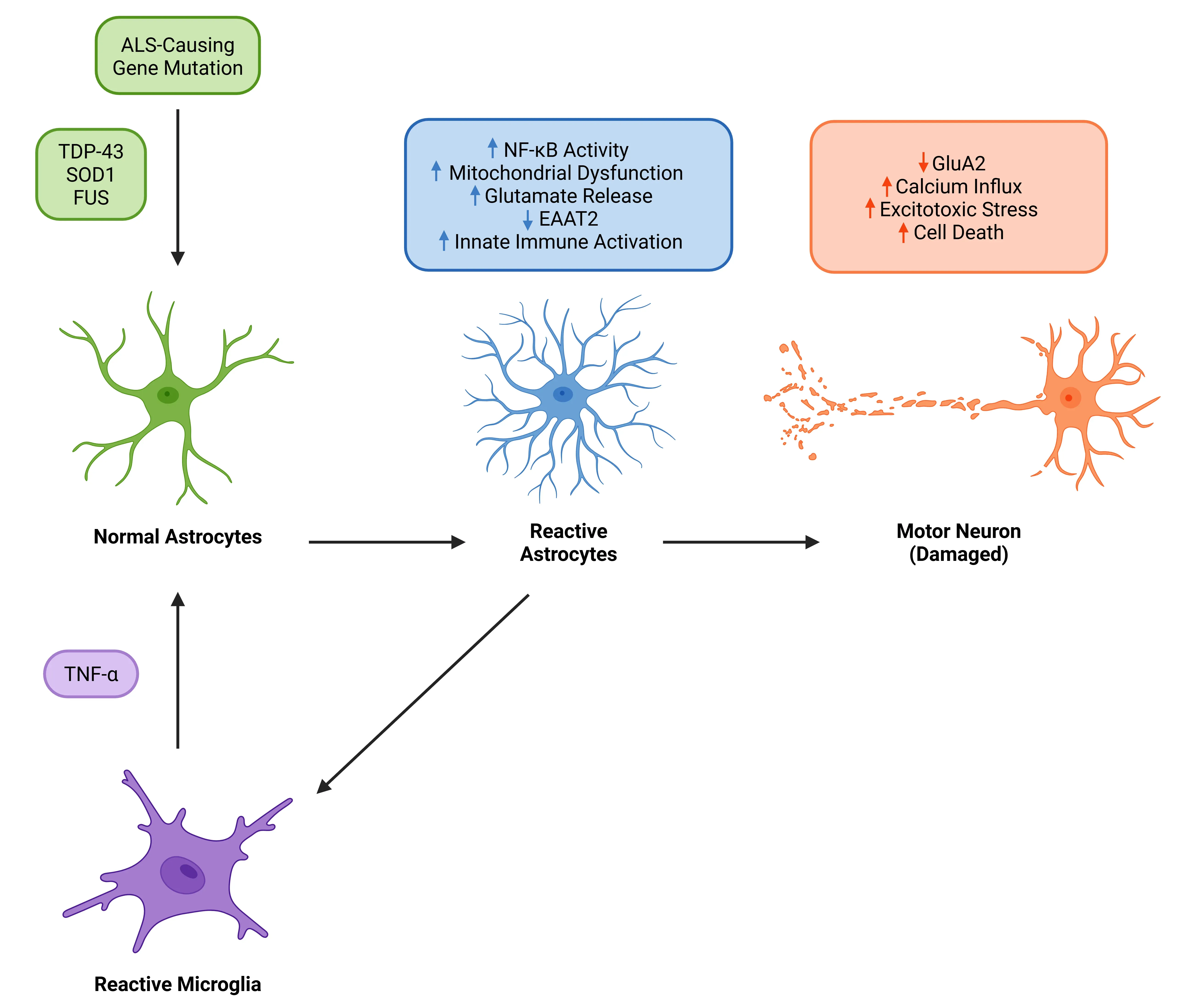
TNF-αは、ALSにおけるアストロサイト反応性と運動ニューロン変性の重要な駆動因子として作用します。 TDP-43、SOD、または FUSの変異に関連するALSでは、反応性ミクログリアから放出されるTNF-αが、TNFRシグナル伝達を介してアストロサイトの反応性を促進します。これによりアストロサイト内でNF-κBが活性化され、グルタミン酸の除去が阻害されます。その結果、興奮毒性ストレスが生じ、運動ニューロンの細胞死を引き起こします。反応性アストロサイトはさらに神経炎症を増幅させ、運動ニューロン喪失を加速させるフィードバックループを形成します。
多発性硬化症(MS)
多発性硬化症(MS)において、アストロサイトはTNF-αの効果を媒介する重要な役割を担い、炎症カスケードと神経毒性および修復プロセスを結びつけています。TNF-αは脳脊髄液(CSF)で一貫して上昇しており、急性および慢性活動性MS病変の両方で検出されます。その高レベルは、より重度の障害および疾患進行の加速と相関しています(Sharief, 1991 ;Kosa, 2022 )。病変レベルの病理学的所見は、急性期および慢性活動性プラークにおけるTNFの上昇を確認しており、TNFシグナル伝達が活動的な組織損傷部位と一致していることを示しています(Mazziotti, 2024 )。
アストロサイトが局所的なTNFシグナル伝達を増幅する主要なメカニズムには、膜結合型TNFのタンパク質分解処理が関与しています。活動性MS病変内では、反応性アストロサイトがADAM17/TACEをアップレギュレートし、これにより膜貫通型TNF(tmTNF)が可溶性型に切断されます。この変化によりTNFR1が優先的に活性化され、アポトーシスおよび炎症促進カスケードが増強されます(Plumb, 2006 )。実験的自己免疫性脳脊髄炎(EAE)において、アストロサイト由来のTNFR1シグナル伝達が海馬シナプス再構築と認知障害を促進することが示され、グリア細胞のTNFシグナル伝達が神経ネットワーク機能障害に直接関与することが明らかとなりました(Habbas, 2015 )。 その後の遺伝子救済研究により、アストロサイト特異的にTNFR1を再導入するだけで、シナプスおよび行動学的EAE表現型を再確立できることが裏付けられました(Di Castro, 2022 )。これらの知見は、アストロサイトが単なる応答者ではなく、TNFを介した神経毒性の増幅器としての役割を担っていることを示唆しています。
同時に、アストロサイトは保護的なTNF経路を活用する能力も有しており、MSにおけるその二重の役割を強調しています。アストロサイトはTNFR1とTNFR2の両方を発現しており、TNFR2シグナル伝達が神経保護と修復を促進することを示す証拠が増えています。脱髄モデルにおいて、アストロサイトにおけるTNFR2の活性化は、pCXCL12によるオリゴデンドロサイト動員などのメカニズムを通じて、抗炎症性表現型を促進し、再髄鞘形成をサポートします(Brambilla, 2011 ;Patel, 2012 )。最近の研究では、TNFR2が星状細胞の炎症促進プログラムを抑制し、髄鞘形成に対する栄養的サポートを強化することが追加で示されており、TNFR1を介した損傷に対するカウンターバランスとしての役割が強調されています(Raphael, 2019 ;Pegoretti, 2023 )。
これらの機序に関する知見は、多発性硬化症(MS)における広範な抗TNF療法の臨床的試みが失敗した理由を説明する助けとなります:
- 非選択的TNF遮断は以下と関連しています:
- 脱髄性病変の逆説的な増悪
- 新規中枢神経系炎症症候群の発生(W. Xie, 2024 )
その代わりに、治療の方向性はTNFシグナル伝達の選択的調節へと移行しています(Brambilla, 2011 ;Pegoretti, 2023 ):
- 前臨床およびトランスレーショナル研究において有望な戦略には以下が含まれます:
- TNFR1の阻害
- sTNFの中和
- TNFR2の温存または作動
これらの知見を総合すると、TNFは多発性硬化症(MS)におけるアストロサイト応答を調整する中核的なサイトカインとして位置づけられ、損傷と修復のバランスを絶えず左右していることがわかります。アストロサイトにおけるTNFシグナル伝達の二重性は、治療標的化における課題と可能性を浮き彫りにしており、受容体選択的調節は神経保護と再髄鞘形成のための魅力的な戦略となります。
アストロサイトにおけるTNF-αシグナル伝達を標的とする治療戦略にはどのようなものがありますか?
現在の治療戦略は、アストロサイトにおけるTNF-αシグナル伝達の異なる側面を標的とする4つの主要なアプローチに分類できます:
TNF-α受容体/リガンド阻害剤
治療戦略は、病的なTNFR1活性を抑制しつつ、TNFR2の修復機能を維持または強化することで、アストロサイトにおけるTNF-αシグナル伝達を微調整することをますます目指しています。このバランスは極めて重要です。なぜなら、TNFR1の選択的阻害やsTNFの中和は、中枢神経系(CNS)内の栄養作用および再髄鞘化プログラムを支えるtmTNF-TNFR2シグナル伝達を温存できるからです(Fischer, 2020 )。
主なアプローチは以下の通りです:
- 受容体選択的拮抗薬
- ヒト一価性TNFR1特異的拮抗薬であるアトロシマブは、EAEモデルを含む炎症モデルにおいて有効性を示しており、選択的TNFR1遮断がアストロサイト中心の介入として有効であることを実証しています。
- 追加の非臨床研究では、これらの効果が急性神経変性にも及ぶことが確認され、この薬剤クラスのより広範な神経保護の可能性が強調されています( 、Ort-Casa、2023 )。
- リガンド標的戦略:
- ドミナントネガティブTNFおよびXPro1595(ペギパネラミン)は、mTNF-TNFR2シグナル伝達を保持しつつsTNFを中和します(MacPherson, 2017 ;De Sousa Rodrigues, 2019 )。
- ADAM17/TACE阻害は、tmTNFがsTNFへ切断されるのを防ぎ、TNFR2経路をさらに促進します(L. Xie, 2024 )。
これらの有望な結果にもかかわらず、ほとんどの生物学的製剤は血液脳関門(BBB)透過性が低いため、臨床応用は依然として困難です。この制限が分子シャトルや中枢神経系(CNS)標的型送達システムの開発を促進してきました(Kouhi, 2021 )。加えて、非選択的全身性TNF阻害剤の臨床経験では、脱髄性疾患の逆説的な悪化が認められており、神経炎症環境における受容体およびリガンド選択的戦略の重要性が強調されています(Mazziotti, 2024 )。
下流シグナル伝達の標的化
別のアプローチとして、TNF受容体の下流にあるシグナル伝達経路、特にアストロサイト反応性を調節するNF-κBおよびMAPKの調節が挙げられます。
- 主要な下流標的:
- NF-κB: アストロサイト特異的活性化は生体内での神経炎症を増幅させるため、選択的阻害が有益である可能性が示唆されています。ただし、新たなヒト細胞研究では、その神経細胞への影響は状況に応じて保護的でも有害でもあり得ることが明らかになっています(Giovannoni, 2020 )。
- p38α MAPK: MW150などの選択的阻害剤は、アルツハイマー病モデルにおいて神経炎症を抑制し認知機能を改善し、臨床応用可能性を示唆しています(Frazier, 2024 )。
- MEK1/2(ERK経路):阻害により、パーキンソン病関連変異を有するアストロサイトにおける微小血管形態形成が回復します。正常状態では、MEK/ERK経路はアストロサイトがストレス応答を調節し、バランスの取れた炎症シグナル伝達を構築するのに役立ちます。しかし、疾患状態ではこの保護的役割が病理学的に増幅され、MEK/ERKが血液脳関門(BBB)安定化のための操作可能なレバーであり、神経変性疾患における有望な治療標的であることを浮き彫りにしています(de Rus Jacquet, 2023 )。
- JNK:その阻害は星状細胞からのCXCL1放出と下流の神経細胞ストレスを抑制し、JNKを並行経路の標的として位置づけています(Zhang, 2022 )。
アストロサイト表現型の調節
補完的な治療戦略は、アストロサイトを神経毒性表現型から恒常性維持または修復状態へと再プログラムすることを目指します。IL-1α/TNF/C1q軸を遮断することで、A1様アストロサイトへの転換を防ぎ、生体内における神経細胞およびオリゴデンドロサイト生存率を維持します。これはアストロサイト表現型の調節が治療的効果を持つことを示す最も説得力のある実証の一つです(Liddelow, 2017 )。その他の有望なメカニズムには以下が含まれます:
- ELOVL1阻害:毒性脂質粒子の放出を阻害し、脂質代謝が薬理学的標的となり得る脆弱性であることを示しています(Guttenplan, 2021 )。
- HDAC3阻害剤:炎症性遺伝子の活性化を抑制しつつ、支持的なアストロサイト機能を温存します(Clayton, 2024 )。
- GLP-1受容体作動薬(例:NLY01)は、ミクログリア誘発性アストロサイト毒性を抑制し、現在アルツハイマー病(AD)およびパーキンソン病(PD)における臨床試験が進行中です(Yun, 2018 ;Park, 2021 )。
遺伝子治療および新規生物学的製剤
遺伝子治療および生物学的製剤は、アストロサイト特異的なTNFシグナル制御の選択肢をさらに拡大しています:
- アストロサイトへのIL-2遺伝子導入は、全身免疫に影響を与えることなく、局所的な制御性T細胞を増加させ、神経炎症を軽減します(Yshii, 2022 )。
- GFAPプロモーター、次世代アストロサイト特異的要素、広範な血清型エンハンサーを用いたAAVベクターの進歩により、げっ歯類および大型動物種双方においてアストロサイト選択的遺伝子導入が可能となりました(O'Carroll, 2021 ;Heffernan, 2022 ;Gleichman, 2023 )。
- TNFR2作動薬投与に続いてTNFR1拮抗薬を投与するといった併用戦略は、ヒト化EAEモデルにおいて治療成績を改善し、受容体活性を時間的に偏向させることで治療効果を最大化しつつリスクを最小化できることを示しています(Pegoretti, 2023 )。
総括しますと、アストロサイトにおけるTNF-αシグナル伝達を標的とすることは、修復と血液脳関門(BBB)の安定性を支える保護機能を維持しつつ、その病理学的炎症反応を抑制する必要性を浮き彫りにしています。現在の治療法における脳内浸透性の低さが依然として主要な障壁ではありますが、選択的阻害剤、送達システム、およびアストロサイトに焦点を当てた介入法の進歩により、TNF-α調節を実用的な神経保護治療へと転換する可能性が高まっています。
TNF-αは神経変性において他のサイトカインとどのように比較されますか?
神経変性疾患において、TNF-αは他のサイトカインとはいくつかの重要な点で異なり、疾患進行におけるその役割と治療標的としての可能性の両方を形作っています。
TNF-αの特異的機能(アポトーシス、ネクロプトーシス、炎症)
TNF-αは、その受容体配線が炎症シグナル伝達とプログラム細胞死を結びつけるため、神経炎症性サイトカインの中で独特の位置を占めています。他のメディエーターとは異なり、TNF-αはTNFR1を介して遺伝子誘導をアポトーシスおよびネクロトーシスに結び付け、NF-κBによる生存および炎症促進出力に加え、RIPK1/RIPK3/MLKL経路を活性化します(Holbrook, 2019 ;van Loo, 2023 )。
これに対し:
- IL-6は以下を介してシグナル伝達を行います:
- 膜結合型IL-6Rを介した「古典的」シグナル伝達:保護的・再生的応答を促進します。
- 可溶性IL-6Rを介した「トランスシグナリング」は、炎症を増幅させます。
- 可溶性IL-6受容体は主にアゴニストとして作用し、可溶性受容体が拮抗作用を示す傾向があるTNFとは対照的です(Rose-John, 2021 )。
- STAT3優位の応答を活性化し、アストロサイトの反応性を増幅させ、血管の安定性を損ない、血液脳関門(BBB)の破壊に寄与します(Mora, 2024 )。
- インターフェロン-γ(IFN-γ)は、アストロサイトを抗原提示へと再プログラムし、TNF誘導性の細胞死促進状態ではなく、インターフェロン応答性反応性アストロサイト(IRRA)を生成します(Rostami, 2020 ;Prakash, 2024 ;Lee, 2023)。
これらの知見は、TNF-αが先天性炎症シグナルと実行者死経路を独自に橋渡しし、IL-6やIFN-γよりも神経変性の「最終共通経路」に近い位置にあることを示唆しています。
アストロサイトにおけるTNF-αと他のサイトカインとの相乗効果および相違点
機能的には、TNF-αは、特に他のメディエーターと共同で作用する場合、アストロサイトを不適応な表現型に備えさせるプライミングサイトカインとして作用することが多いです。
主な相互作用には以下が含まれます:
- TNF-α + IL-1α + C1q:アストロサイトをA1様状態へと誘導し、シナプス支持機能の喪失、ならびにニューロンおよびオリゴデンドロサイトの死を引き起こします(Liddelow, 2017 )。
- TNF-α + IL-β:ヒトiPS細胞由来アストロサイトにおいてNF-κBシグナル伝達を強力に活性化し、その構造を再構築するとともに強力な免疫反応性状態を誘導します(Hyvärinen, 2019 )。
これらの知見は、TNF-αがIL-1α(C1qと共に)およびIL-1βと相乗的に作用して神経毒性および免疫反応性アストロサイト状態を誘導する一方で、IFN-γやIL-6などのサイトカインは異なる経路を活性化し、疾患におけるアストロサイトの挙動を形作るサイトカインプログラムの差異を強調していることを示しています。
TNF駆動型アストロサイトの空間的・単一細胞シグネチャ
単一細胞トランスクリプトーム解析および空間トランスクリプトーム解析により、TNFα応答性アストロサイトが疾患に関連する独自の状態を形成することが示されました:
- アルツハイマー病(AD)において:
- snRNA-seq解析により、プラーク隣接アストロサイトは正常な支持的遺伝子プログラムを失い、代わりにTNFによって駆動される補体および炎症経路を活性化することが示されました(Dai, 2023 )。
- 空間トランスクリプトミクスにより、これらのプラーク境界アストロサイトがTNF/IL-1シグナル伝達によって強く形作られ、変性するニューロン周辺にクラスターを形成していることがさらに確認されました(He, 2024 )。
- メタ解析により、TNF-α、IL-6、IL-1βがアルツハイマー病の進行に伴い並行して増加し、認知機能低下の悪化と直接相関することが示されています(Lista, 2024 ;Serna, 2025 )。
- パーキンソン病(PD)においては:
- スモールRNAシーケンス解析により、グリア細胞の広範な活性化が確認されました。これには黒質において慢性的なTNF/IL-1駆動型炎症を反映する遺伝子発現を示すCD44高発現アストロサイトの一群も含まれます(Smajić, 2022 )。
TNF-αと他のサイトカインの治療的選択性
TNF-αは、そのシグナル伝達機構が有害なTNFR1経路を選択的に標的としつつ、保護的なTNFR2機能を温存することを可能にするため、治療的に際立っています。この種の選択的バランスはTNFに特有であり、IL-6やIFN-γの遮断では達成できません(Fischer, 2020 ;Papazian, 2021 )。
- 非選択的抗TNF療法は中枢神経系(CNS)の自己免疫を悪化させる可能性があります(Mazziotti, 2024 )。
- IL-6療法(例:sgp130Fc)は炎症を阻害しつつ保護的な「古典的」IL-6シグナル伝達を温存しますが、TNF受容体シグナル伝達に特有の細胞死経路には作用しません(Rose-John, 2021 )。
- IFN-γ療法は、TNFによって駆動されるアポトーシスおよびネクロトーシスのカスケードではなく、抗原提示などの免疫関連アストロサイト機能を調節します(Prakash, 2024 ;Lee, 2023 )。
これらの比較から、TNF-αはサイトカインの中でも特異的であることが示されています。そのシグナル伝達は治療的に微調整が可能であり、TNFR1またはsTNFを選択的に遮断することで神経毒性および細胞死経路を抑制しつつ、TNFR2を介した修復機能を維持できるからです。これはIL-6やIFN-γによる介入では達成できない治療的バランスです。
総括しますと、TNF-αは他のサイトカインよりも神経変性進行に直接的に関連する形でアストロサイトの挙動を形作るという証拠が示されています。炎症性シグナルを細胞死経路に結合させ、不適応なアストロサイト状態を増幅し、領域特異的な転写変化を駆動することにより、TNF-αはアストロサイトを疾患メカニズムと治療機会の両方の中心に位置づけています。
当チームは、アストロサイトにおけるTNF-αの機能や神経変性疾患への関与に関するご質問、ならびに治療効果研究に使用しているアルツハイマー病(AD)、筋萎縮性側索硬化症(ALS)、パーキンソン病(PD)モデルに関する具体的な情報について、喜んでご説明いたします。
当社の神経変性疾患モデルについて、さらに詳しくご覧ください
関連コンテンツ
神経変性疾患におけるTNF-αとミクログリアに関する最新情報、ならびに神経変性疾患の動物モデルにおける治療薬評価に関連するベストプラクティスについて。
神経変性疾患におけるTNF-αとミクログリア
ミクログリアにおける腫瘍壊死因子-α(TNF-α)の機能と、神経変性進行への寄与についての概要。
IL-1βとは何ですか?
IL-1βの概要、全身性疾患および神経疾患におけるその炎症誘発作用、ならびにIL-1β拮抗作用を基盤とした治療戦略について概説します。
ミトコンドリア機能障害とミクログリアおよびアストロサイト
アルツハイマー病、パーキンソン病、ALSを含む神経変性疾患におけるミトコンドリア機能障害のミクログリアおよびアストロサイトにおける役割。
パーキンソン病におけるミクログリア、アストロサイト、およびα-シヌクレイン
α-シヌクレインがパーキンソン病およびその他のシヌクレイン病においてミクログリアおよびアストロサイトに及ぼす影響。
インターロイキン-1ベータ(IL-1β)と神経変性疾患
アルツハイマー病(AD)、パーキンソン病(PD)、筋萎縮性側索硬化症(ALS)などの神経変性疾患におけるIL-1βの役割。
パーキンソン病におけるミクログリア、アストロサイト、およびα-シヌクレイン
α-シヌクレインがパーキンソン病およびその他のシヌクレイン病においてミクログリアおよびアストロサイトに及ぼす影響。