Le TNF-α et les astrocytes dans les maladies neurodégénératives
Présentation générale de la signalisation du TNF-α dans les astrocytes, de son rôle dans la neurodégénérescence et des stratégies thérapeutiques ciblant cette voie.
Cette ressource décrit:
- Comment la signalisation du TNF-alpha affecte-t-elle la fonction des astrocytes dans les maladies neurodégénératives?
- Quels rôles les astrocytes réactifs jouent-ils dans la neuroinflammation médiée par le TNF-alpha?
- Comment le TNF-alpha contribue-t-il à la progression des maladies neurodégénératives par l'intermédiaire des astrocytes?
- Quelles sont les stratégies thérapeutiques qui ciblent la signalisation du TNF-alpha dans les astrocytes?
- Comment le TNF-α se compare-t-il aux autres cytokines dans la neurodégénérescence?
Comment la signalisation du TNF-α affecte-t-elle la fonction des astrocytes dans les maladies neurodégénératives?
Les astrocytes réagissent au facteur de nécrose tumorale alpha (TNF-α) par le biais de deux systèmes récepteurs, TNFR1 et TNFR2, dont les activités opposées établissent l'équilibre entre neurotoxicité et réparation. L'activation du TNFR1 dans les astrocytes suffit à induire un dysfonctionnement synaptique et des déficits de mémoire in vivo, comme le montre le modèle expérimental d'encéphalomyélite auto-immune (EAE), où les troubles de l'hippocampe sont directement liés à la signalisation gliale du TNF (Habbas, 2015 ). Dans les modèles précliniques de stress chronique, tels que les souris soumises à un stress chronique imprévisible et léger (CUMS), la régulation à la hausse du TNFR1 est liée à des symptômes de type dépressif marqués par une astrogliose et une apoptose neuronale, tandis que le blocage du TNFR1, par voie pharmacologique ou génétique, inverse ces effets (Gao, 2024 ). En revanche, la signalisation TNFR2 dans les astrocytes limite l'astrogliose réactive et favorise la remyélinisation en supprimant les programmes pro-inflammatoires dans les conditions de démyélinisation, tout en soutenant la fonction synaptique, la plasticité et la cognition de l'hippocampe dans des états physiologiques (Raphael, 2019 ; Carney, 2025).
Cette divergence des récepteurs crée des opportunités thérapeutiques:
- La neutralisation sélective du TNF soluble (sTNF), qui active principalement le TNFR1, améliore la remyélinisation dans les modèles de démyélinisation (par exemple, le modèle de démyélinisation à la cuprizone), tout en préservant la signalisation bénéfique via la voie transmembranaire TNF (tmTNF)-TNFR2 (Karamita, 2017).
- Des agonistes sélectifs du TNFR2 sont en cours de développement afin d'exploiter cette divergence tout en évitant les complications liées au blocage non sélectif du TNF (Pegoretti, 2023).
Ensemble, ces résultats mettent en évidence le rôle du TNFR1 et du TNFR2 en tant que commutateur moléculaire dans les astrocytes, faisant pencher la balance entre les programmes neurotoxiques et réparateurs, et soulignent le ciblage sélectif des récepteurs comme une voie thérapeutique prometteuse.
Au-delà des effets spécifiques aux récepteurs, le TNF-α orchestre les transitions d'état astrocytaire qui font basculer le paysage glial entre les programmes protecteurs et neurotoxiques. Un exemple frappant est la triade de cytokines IL-1β, TNF-α et C1q, qui fait passer les astrocytes homéostatiques à des états A1 neurotoxiques caractérisés par l'expression du complément C3. La perturbation de cette voie empêche la conversion A1 et prolonge la survie dans les modèles de SLA, reliant directement la signalisation TNF astrocytaire à la progression de la maladie (Liddelow, 2017 ; Guttenplan, 2020). Cependant, ce phénotype n'est pas universellement nocif ; dans les maladies à prions, l'ablation des astrocytes C3+ aggrave la dégénérescence, soulignant les rôles spécifiques à la maladie et au contexte (Hartmann, 2019 ). Les astrocytes dérivés d'iPSC humaines reproduisent cette dynamique, où le TNF-α, seul ou associé à l'IL-1β, déclenche l'activation du NF-κB et la régulation à la hausse du C3, altérant fonctionnellement la clairance du glutamate (Hyvärinen, 2019). Un profilage multi-omique récent montre que les astrocytes de type A1 dans les tissus cérébraux humains atteints de MA et de SLA se regroupent autour des sites de perte neuronale, renforçant ainsi leur potentiel pathogène (Escartin, 2021). Dans la maladie de Parkinson (MP), les astrocytes exposés au TNF-α par opposition aux fibrilles d'α-synucléine suivent des schémas réactifs différents, mais convergent tous deux vers un dysfonctionnement mitochondrial, un point final commun qui contribue à la progression de la maladie (Russ, 2021). Ensemble, ces résultats montrent que le TNF-α est un facteur clé des transitions de l'état astrocytaire, les poussant vers des programmes neurotoxiques qui influencent la maladie de manière dépendante du contexte.
Pour une revue sur la fonction du TNF-α dans les microglies et sa contribution aux maladies neurodégénératives, voir : TNF-α et microglies dans les maladies neurodégénératives
Au-delà de la formation du phénotype astrocytaire, le TNF-α modifie directement les fonctions astrocytaires clés qui sont essentielles à la santé neuronale et vasculaire :
- Le TNF-α réduit l'absorption du glutamate en régulant à la baisse l'EAAT2/GLT-1 par le biais d'une voie TNFR1/NF-κB, augmentant ainsi le stress excitotoxique sur les motoneurones (Jiang, 2019).
- Dans les astrocytes dérivés d'iPSC humaines, le TNF-α et d'autres cytokines pro-inflammatoires diminuent la clairance du glutamate et renforcent les signatures immunitaires réactives (Hyvärinen, 2019).
- Le TNF-β renforce la gliotransmission. Dans les circuits cérébelleux, l'activation du TNFR1 dans les cellules gliales de Bergmann augmente la libération de glutamate et modifie l'excitabilité neuronale par le biais de mécanismes dépendants du mGluR1 (Shim, 2018).
- Un axe TNF-STAT3 dans des conditions inflammatoires compromet l'intégrité de la barrière hémato-encéphalique (BHE), et les astrocytes PD sensibilisés à l'inflammation ne parviennent pas à soutenir la morphogenèse microvasculaire - un défaut inversé par l'inhibition de MEK1/2 (de Rus Jacquet, 2023).
Ces résultats démontrent que le TNF-α non seulement entraîne des changements de l'état inflammatoire, mais compromet également les rôles essentiels des astrocytes dans l'homéostasie du glutamate, la communication neurone-glie et la stabilité vasculaire, accélérant ainsi les processus neurodégénératifs.
Ces résultats divers s'expliquent par les réseaux de signalisation en aval des récepteurs du TNF, qui couplent les centres inflammatoires et métaboliques au contrôle de l'état astrocytaire:
- L'activation du NF-κB (RelA/p65) entraîne des programmes transcriptionnels inflammatoires et contribue au dysfonctionnement mitochondrial, ces déficits bioénergétiques étant confirmés dans des modèles d'astrocytes humains (Russ, 2021).
- Les modules de signalisation MAPK croisent également les voies TNF: l'activité ERK/MAPK régule les fonctions de soutien de la BHE dans les astrocytes PD.
- L'activation de JNK en aval du TNF microglial induit la production astrocytaire de CXCL1 qui remodèle la communication neurone-glie in vivo (Zhang, 2022; de Rus Jacquet, 2023).
Dans l'ensemble, la dichotomie entre TNFR1 et TNFR2, intégrée aux hubs NF-κB et MAPK, positionne le TNF-α comme un régulateur principal des transitions d'état des astrocytes, orchestrant des programmes neurotoxiques ou réparateurs dans les maladies neurodégénératives (Raphael, 2019).
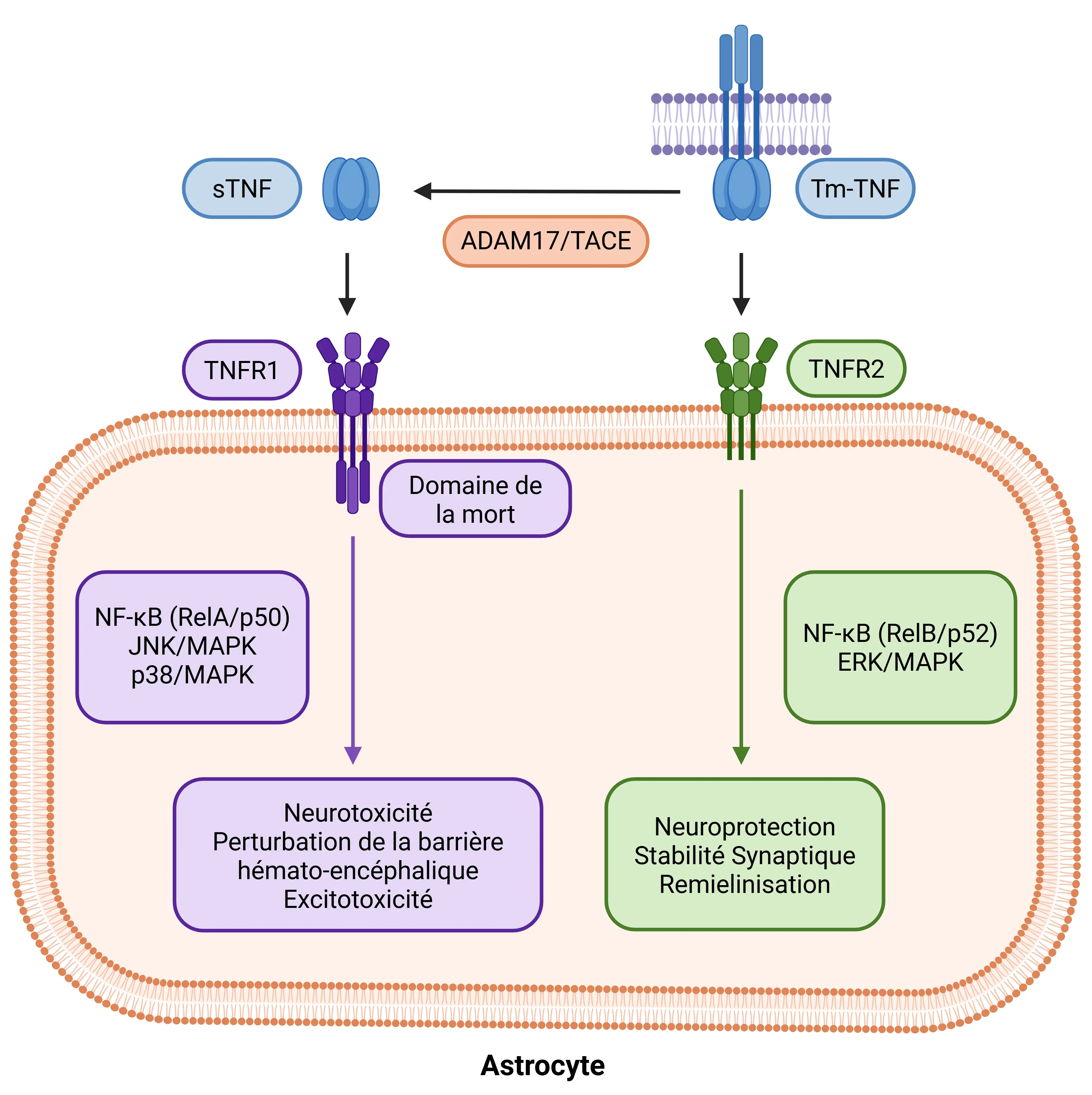
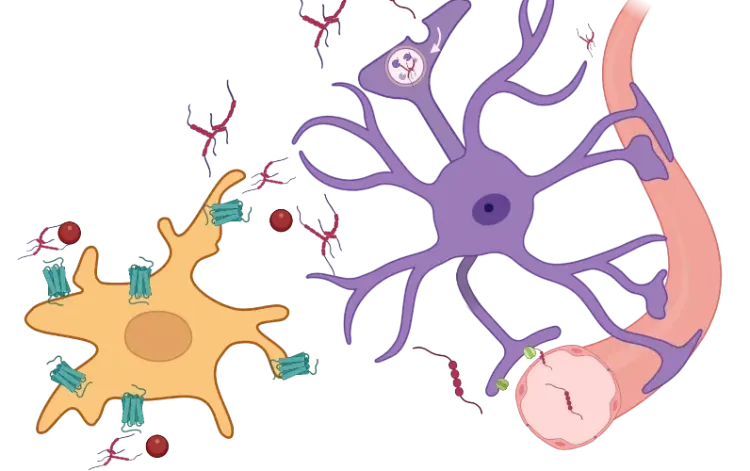
Dichotomie du récepteur TNF-α dans les astrocytes. Les réponses astrocytaires au TNF-α dépendent de la forme du ligand et de la spécificité du récepteur. Le sTNF clivé du tmTNF par ADAM17/TACE active le TNFR1, déclenchant les voies NF-κB (RelA/p50), JNK et p38 MAPK qui entraînent une neurotoxicité et une perturbation de la BHE. En revanche, le tmTNF se lie préférentiellement au TNFR2, signalant via NF-κB non canonique (RelB/p52) et ERK/MAPK pour favoriser la neuroprotection, la stabilité synaptique et la remyélinisation. Cette dichotomie positionne le TNF-α comme un régulateur clé de l'état astrocytaire entre la lésion et la réparation.
Quel rôle jouent les astrocytes réactifs dans la neuroinflammation médiée par le TNF-α?
Les astrocytes réactifs sous l'influence du TNF-α adoptent plusieurs rôles néfastes qui vont au-delà de la simple activation gliale. Nous décrivons ici comment ces états contribuent à la neuroinflammation, au dysfonctionnement vasculaire et aux lésions neuronales directes dans différents contextes pathologiques.
Toxicité gliale et dysfonctionnement vasculaire
La réactivité astrocytaire est profondément influencée par le TNF-α, qui agit comme un amplificateur clé de la communication entre les microglies et les astrocytes dans le cerveau enflammé. Le TNF-α dérivé des microglies, associé à l'IL-1α et au C1q, suffit à convertir les astrocytes homéostatiques en états A1 neurotoxiques in vitro et in vivo, établissant ainsi le TNF-α comme un inducteur clé de l'astrogliose. La neutralisation de cette triade IL-1α/TNF/C1q empêche la conversion A1 et préserve la survie des neurones et des oligodendrocytes, confirmant que la réactivité dépendante du TNF-α est une cause de pathologie plutôt qu'un simple sous-produit de la maladie (Liddelow, 2017). Dans les circuits hippocampiques, l'activation du TNFR1 astrocytaire par le TNF-α déclenche une cascade astrocytaire-neuronale qui remodèle les synapses excitatoires et altère la mémoire contextuelle (Habbas, 2015).
Au niveau de l'interface vasculaire, le TNF entraîne les astrocytes dans un état inflammatoire réactif dépendant de STAT3, les amenant à libérer des facteurs qui perturbent l'intégrité de la BHE et induisent une inflammation endothéliale vasculaire (Kim, 2022). S'appuyant sur ces preuves de perturbation vasculaire, des études récentes sur des puces cérébrales humaines démontrent que les astrocytes activés par le TNF non seulement déstabilisent la fonction de barrière, mais altèrent également la morphogenèse microvasculaire, un défaut réversible par l'inhibition de MEK1/2 (de Rus Jacquet, 2023).
Dans l'ensemble, les astrocytes réactifs au TNF-α agissent comme des facteurs clés de dysfonctionnement en alimentant des états gliaux toxiques, en perturbant la communication synaptique et en affaiblissant l'intégrité de la BHE et des microvaisseaux, amplifiant ainsi les dommages neuroinflammatoires.
Facteurs inflammatoires et oxydatifs de la neuroinflammation
Une fois que les astrocytes adoptent un état réactif induit par le TNF-α, ils deviennent des sources actives d'inflammation et de stress oxydatif qui propagent les dommages bien au-delà du compartiment glial. Un mécanisme central est l'activation de la voie NF-κB, qui induit la production de médiateurs pro-inflammatoires :
Ces signaux recrutent des leucocytes et amplifient l'activité immunitaire locale du SNC (Giovannoni, 2020).
En parallèle, les astrocytes réactifs produisent des espèces réactives de l'oxygène et de l'azote (ROS/NOS), reliant la signalisation du TNF-α aux voies du stress oxydatif qui déstabilisent davantage l'homéostasie neuronale et vasculaire (Ding, 2021). L'interaction bidirectionnelle avec les microglies intensifie ces réponses : l'activation de NF-κB par les astrocytes suffit à stimuler la prolifération microgliale et l'infiltration leucocytaire, ce qui montre que les astrocytes aggravent activement la neuroinflammation plutôt que d'y répondre passivement (Ouali Alami, 2018).
Voir : Dysfonctionnement mitochondrial dans les microglies et les astrocytes
Les modèles d'astrocytes dérivés d'iPSC humaines reproduisent ces caractéristiques, car la stimulation combinée de l'IL-1β et du TNF-α induit la translocation nucléaire du NF-κB, un remodelage morphologique et l'acquisition d'un phénotype immunoréactif (Hyvärinen, 2019). Conformément à ces modèles expérimentaux, le séquençage d'ARN à noyau unique (snRNA-seq) provenant de tissus humains atteints de MA et de SEP démontre que les états astrocytaires induits par NF-κB se regroupent autour des plaques et des lésions, soulignant leur pertinence clinique directe (Escartin, 2021).
Ensemble, ces résultats montrent que les astrocytes réactifs au TNF-α agissent comme de puissants moteurs de l'inflammation et du stress oxydatif, amplifiant activement les cascades neuroinflammatoires et aggravant la vulnérabilité neuronale et vasculaire.
Neurotoxicité et dysfonctionnement des circuits
La réactivité des astrocytes induite par le TNF-α non seulement les prive de leurs rôles de soutien normaux, mais en fait également des sources actives de lésions neuronales et oligodendrogliales. Sur le plan fonctionnel, les astrocytes A1 perdent leur soutien pro-synaptogène normal et sécrètent des facteurs qui détruisent les neurones et les oligodendrocytes, reliant directement les états réactifs induits par le TNF-α à la neurodégénérescence (Liddelow, 2017). L'un des mécanismes implique la sécrétion de lipides saturés dans les lipoparticules APOE/APOJ; le blocage de l'enzyme ELOVL1, qui métabolise les lipides, empêche cette toxicité, reliant ainsi le métabolisme lipidique des astrocytes à la mort neuronale induite par le TNF (Guttenplan, 2021).
Au niveau des circuits, une exposition prolongée au TNF-α, seul ou associé à l'IL-6, perturbe les schémas de décharge neuronale dans les co-cultures de neurones et d'astrocytes humains, entraînant des rythmes anormaux qui pourraient être à l'origine des déficits cognitifs et comportementaux observés lors d'une inflammation (Goshi, 2025). Des perturbations similaires ont été observées in vivo, où une exposition chronique au TNF-α entraîne une hyperexcitabilité de l'hippocampe et une activité épileptiforme (Vezzani, 2020).
Dans l'ensemble, les astrocytes réactifs au TNF-α apparaissent comme des facteurs déterminants de la perte neuronale et de l'instabilité du réseau, reliant directement la réactivité gliale à la progression des maladies neurodégénératives.
Ensemble, ces preuves démontrent que le TNF-α remodèle fondamentalement la biologie des astrocytes, les faisant passer du statut de gardiens de l'homéostasie du SNC à celui de moteurs centraux du dysfonctionnement. En plaçant les astrocytes au carrefour de l'inflammation, de la compromission vasculaire et de la vulnérabilité neuronale, le TNF-α les révèle non pas comme des répondeurs passifs, mais comme des régulateurs décisifs de la progression neurodégénérative. Cette perspective redéfinit les astrocytes, qui passent du statut d'acteurs secondaires à celui de cibles thérapeutiques essentielles dans la lutte contre la neuroinflammation chronique.
Comment le TNF-α contribue-t-il à la progression des maladies neurodégénératives par le biais des astrocytes?
Une exposition chronique au TNF-α pousse progressivement les astrocytes vers des états néfastes qui modifient leur expression génétique, leur structure et leur métabolisme. Même à de faibles niveaux, la signalisation persistante du TNF-α provoque un élargissement des astrocytes, une réorganisation des gènes inflammatoires et un stress métabolique, ce qui conduit les chercheurs à considérer la réactivité des astrocytes non pas simplement comme « protectrice ou toxique », mais comme un spectre d'états changeants (Escartin, 2021).
Les signaux inflammatoires microgliaux contenant du TNF-α renforcent la transition vers des astrocytes de type A1 neurotoxiques, établissant un lien direct entre l'activation chronique du système immunitaire inné et le dysfonctionnement des astrocytes dans les maladies neurodégénératives (Liddelow, 2017). Les astrocytes dérivés d'iPSC humaines reproduisent ce processus, dans lequel le TNF-α (± IL-1β) active rapidement le NF-κB et remodèle la morphologie et les réseaux de gènes inflammatoires (Hyvärinen, 2019). Dans les astrocytes humains liés à la maladie de Parkinson, le TNF-α ou les fibrilles d'α-synucléine entraînent les astrocytes dans des états immunitaires et métaboliques qui se chevauchent, caractérisés par une respiration mitochondriale défectueuse, montrant ainsi comment une exposition prolongée aux cytokines compromet l'équilibre énergétique cellulaire (Russ, 2021).
Ensemble, ces résultats montrent que la signalisation chronique du TNF-α impose une charge inflammatoire et métabolique persistante aux astrocytes, les faisant basculer dans des états dysfonctionnels qui affaiblissent leurs rôles de soutien et contribuent activement à la progression des maladies neurodégénératives.
Au niveau synaptique, le TNF-α modifie le traitement du glutamate par les astrocytes et la gliotransmission d'une manière qui déstabilise les circuits neuronaux et augmente la vulnérabilité à l'excitotoxicité:
- L'activation du TNFR1 astrocytaire à elle seule peut altérer la transmission excitatrice et nuire à la mémoire hippocampique dans la démyélinisation auto-immune, démontrant ainsi le rôle causal direct du TNF-α astrocytaire dans le dysfonctionnement du réseau (Habbas, 2015).
- Le TNF-α, via la voie TNFR1-NF-κB, réduit l'expression des transporteurs EAAT2/GLT-1, affaiblissant l'absorption du glutamate et exposant les motoneurones à une surstimulation toxique (Jiang, 2019).
- Les astrocytes dérivés d'iPSC humaines co-stimulés par le TNF-α et l'IL-1β présentent une clairance du glutamate altérée et une activation inflammatoire plus forte (Hyvärinen, 2019).
- Le TNF-α améliore la gliotransmission dans les circuits cérébelleux en augmentant la libération de glutamate par les cellules gliales de Bergmann, ce qui augmente l'excitabilité des cellules de Purkinje par le biais de la signalisation mGluR1, déstabilisant ainsi les schémas de décharge (Shim, 2018).
- Une exposition prolongée au TNF-α (± IL-6) perturbe la dynamique de décharge neuronale dans les co-cultures de neurones et d'astrocytes humains sur des réseaux de microélectrodes (MEA), ce qui indique que les cytokines chroniques entraînent des dysrythmies émergentes dans les circuits (Goshi, 2025).
- Des étudesin vivo montrent que la surexpression du TNF-α augmente la susceptibilité aux crises et accélère le déclin cognitif, reliant le dysfonctionnement astrocytaire à une instabilité plus large du réseau (Vezzani, 2020).
Ensemble, ces résultats démontrent que le dysfonctionnement astrocytaire induit par le TNF-α compromet la régulation du glutamate et l'équilibre synaptique, entraînant une instabilité généralisée des circuits et une détérioration cognitive progressive.
Au sein de l'unité neurovasculaire (NVU), les astrocytes activés par le TNF-α transmettent des signaux inflammatoires qui affaiblissent l'intégrité de la BHE et altèrent la fonction vasculaire. Dans les modèles humains et ex vivo, le TNF-α conduit les astrocytes à un état réactif dépendant de STAT3, caractérisé par une régulation à la hausse de SERPINA3 (également connu sous le nom d'α-antichymotrypsine, un inhibiteur de la sérine protéase en phase aiguë), qui réduit l'intégrité de la BHE et favorise le dysfonctionnement endothélial (Kim, 2022).
Les astrocytes porteurs de la mutation LRRK2 liée à la maladie de Parkinson adoptent des profils pro-inflammatoires et ne parviennent pas à soutenir la morphogenèse microvasculaire dans un système BHE de puce cérébrale humaine ; ce déficit est inversé par l'inhibition de MEK1/2 (de Rus Jacquet, 2023). Ces résultats concordent avec des preuves plus générales selon lesquelles les pieds terminaux des astrocytes et les molécules qu'ils sécrètent régulent la stabilité de la BHE, et que le remodelage inflammatoire de ces interfaces contribue au dysfonctionnement vasculaire dans la neurodégénérescence (Yue, 2023). Conformément à ces connaissances mécanistiques, la transcriptomique spatiale des lésions humaines de la sclérose en plaques démontre que les astrocytes activés par le TNF se regroupent dans les régions périvasculaires, produisant des niches riches en cytokines qui déstabilisent activement la structure de la BHE (Mazziotti, 2024).
Ces résultats démontrent que les astrocytes activés par le TNF-α perturbent activement l'intégrité de la BHE et la santé vasculaire, ce qui en fait des contributeurs clés au dysfonctionnement neurovasculaire dans les maladies neurodégénératives.
Dans les principales maladies neurodégénératives, les programmes astrocytaires induits par le TNF-α agissent comme des amplificateurs communs de la progression de la maladie. En couplant la signalisation inflammatoire au dysfonctionnement synaptique, métabolique et vasculaire, les astrocytes transforment les signaux du TNF-α en mécanismes locaux qui accélèrent la vulnérabilité neuronale et le déclin tissulaire. Si ces processus fondamentaux sont communs à tous les troubles, la manière dont les astrocytes réactifs au TNF-α interagissent avec les environnements spécifiques à chaque maladie varie considérablement. Dans les sections suivantes, nous décrivons comment la signalisation du TNF-α par les astrocytes contribue à la progression de la maladie d'Alzheimer, de la maladie de Parkinson, de la SLA et de la sclérose en plaques (SEP).
Maladie d'Alzheimer (MA)
Le TNF-α est apparu comme un médiateur essentiel dans la MA, reliant la pathologie amyloïde β, la neuroinflammation et le dysfonctionnement synaptique. Des taux élevés de TNF-α ont été régulièrement signalés dans le liquide céphalo-rachidien (LCR) et le sérum de patients atteints de troubles cognitifs légers (TCL) et de MA, où des concentrations de base plus élevées prédisent un déclin cognitif plus rapide et un risque accru de progression vers la démence (Kim, 2017; Lista, 2024). Il est important de noter que ces changements biochimiques s'accompagnent d'altérations astrocytaires. Les biomarqueurs des astrocytes réactifs, notamment le GFAP et le YKL-40 dans le LCR, médient les effets de l'amyloïde β et de la protéine tau sur l'atrophie de l'hippocampe et la cognition, établissant un lien entre la réactivité des astrocytes et la neurodégénérescence en aval chez les êtres humains vivants (Ferrari-Souza, 2022; Pelkmans, 2024). Des études neuropathologiques révèlent en outre que les astrocytes réactifs se regroupent autour des plaques Aβ et des enchevêtrements neurofibrillaires, pénétrant au cœur des plaques et contribuant à la fois au confinement et à la propagation de la pathologie (Perez-Nievas, 2018).
Pour une analyse de la morphologie des astrocytes dans le microenvironnement des plaques amyloïdes β, voir: Astrocytes et amyloïde β - Modèles murins de la maladie d'Alzheimer
L'un des mécanismes centraux par lesquels le TNF-α contribue à la MA implique sa capacité à stimuler le processus amyloïdogène dans les astrocytes. Plus précisément, la signalisation du TNF-α:
- Régule à la hausse l'expression de la protéine précurseur amyloïde (APP) et de la BACE1, accélérant ainsi la génération de β et l'accumulation de plaques (Zhao, 2011).
- Perturbe l'homéostasie synaptique en modifiant l'équilibre excitateur-inhibiteur
- Renforce la transmission glutamatergique
- Réduit l'inhibition GABAergique
- Augmente le trafic de surface des récepteurs AMPA,
Ces effets prédisposent collectivement les circuits hippocampiques à l'excitotoxicité (Pribiag, 2013; Heir, 2020). Cette lésion excitotoxique est encore amplifiée par la libération de glutamate médiée par les astrocytes, qui compromet la survie des neurones (Santello, 2011).
Les astrocytes eux-mêmes ne sont pas des répondeurs passifs, mais des moteurs clés de la signalisation TNF-α dans la MA. Des preuves expérimentales montrent que:
- La signalisation du TNF-α persiste dans les cultures hippocampiques dépourvues de microglies, mais est supprimée avec la délétion du TNF spécifique aux astrocytes, ce qui établit les astrocytes comme indispensables à la libération du TNF dépendante de l'activité (Heir, 2024).
- L'activation de NF-κB par les astrocytes régule la production de TNF en réponse à la libération de glutamate, plaçant les astrocytes au centre du remodelage des circuits induit par les cytokines.
- La modulation de la signalisation NF-κB des astrocytes modifie les résultats neuronaux et synaptiques dans les modèles de MA (Jong Huat, 2024).
- Les altérations du traitement du glutamate par les astrocytes, en particulier la réduction de la fonction du transporteur EAAT2/GLT-1, convergent avec l'excitotoxicité induite par le TNF pour exacerber la neurodégénérescence (Wood, 2022).
Les études génétiques renforcent le rôle pathogène du TNF-α. Le polymorphisme G-308A dans le promoteur du TNF-α augmente l'activité transcriptionnelle et l'expression des protéines, plusieurs méta-analyses le reliant à une susceptibilité accrue à la MA (Wang, 2015). Cette variante pourrait agir en synergie avec le génotype APOE-ε4 pour accélérer le déclenchement de l'inflammation et la progression de la maladie (Contreras, 2020), soulignant l'importance de la signalisation du TNF comme dénominateur commun entre la MA sporadique et la MA familiale.
Les études thérapeutiques soulignent le potentiel translationnel du ciblage du TNF-α. Les données montrent que:
- Les patients atteints de maladies inflammatoires systémiques traités par des inhibiteurs du TNF, tels que l'étanercept ou l'adalimumab, présentent une incidence réduite de démence par rapport aux populations non traitées.
- De petits essais pilotes et des rapports de cas utilisant l'administration périspinale d'étanercept font état d'améliorations cognitives rapides et parfois durables chez les patients atteints de MA.
- Des essais contrôlés randomisés à plus grande échelle avec l'étanercept sous-cutané n'ont pas démontré de bénéfices significatifs, probablement en raison de la faible pénétration de la BHE par les produits biologiques de grande taille.
- Les études précliniques avec des inhibiteurs du TNF pénétrant la BHE montrent une réduction de la pathologie Aβ et tau ainsi que des améliorations cognitives dans des modèles murins de MA, renforçant ainsi le bien-fondé d'un traitement ciblant le TNF (Plantone, 2024).
Dans l'ensemble, ces résultats démontrent que le TNF-α, par le biais de mécanismes induits par les astrocytes, est un facteur central de l'accumulation d'amyloïde β, de l'excitotoxicité et de la neurodégénérescence dans la MA, ce qui en fait une cible thérapeutique biologiquement plausible. Bien que les données cliniques restent préliminaires, en particulier en ce qui concerne l'administration à travers la BHE, l'inhibition du TNF-α représente une voie prometteuse pour la modification de la maladie.
Maladie de Parkinson (MP)
Le TNF-α astrocytaire joue un rôle central dans la maladie de Parkinson (MP), apparaissant comme un médiateur qui relie la pathologie de l'α-synucléine à l'amplification neuroinflammatoire et à la vulnérabilité neuronale. Chez les patients atteints de la MP, le TNF-α et son récepteur soluble sont systématiquement élevés dans le cerveau, le LCR et le sang, ce qui est corrélé à la neurodégénérescence dopaminergique, au déclin cognitif et à la gravité globale de la maladie (Liu, 2022). L'accumulation d'alpha-synucléine dans les astrocytes perturbe non seulement le fonctionnement des mitochondries et du réticulum endoplasmique, mais entraîne également un phénotype réactif et pro-inflammatoire (Wang, 2021). Il est important de noter que les astrocytes contenant des inclusions d'alpha-synucléine sécrètent des niveaux élevés de TNF-α, ce qui positionne cette cytokine comme un effecteur central reliant la protéinopathie à l'amplification inflammatoire (Lee, 2010).
Le TNF-α dérivé des astrocytes contribue à une boucle d'auto-alimentation de la réactivité gliale dans la MP.
Pour une analyse approfondie de l'influence de l'alpha-synucléine sur les microglies et les astrocytes dans la maladie de Parkinson et d'autres synucléinopathies, veuillez consulter: Microglies, astrocytes et alpha-synucléine dans la maladie de Parkinson
Avec l'IL-1β et l'IL-6, le TNF-α astrocytaire:
- Amplifie l'activation microgliale, accélérant ainsi la perte neuronale dopaminergique.
- Exacerbe la vulnérabilité neuronale.
- Établit une boucle de rétroaction positive de la réactivité gliale reliant la signalisation des cytokines induite par les astrocytes à la dégénérescence nigrostriatale progressive (Wang, 2023).
Les modèles expérimentaux de la maladie de Parkinson montrent que l'IFN-γ et le TNF-α sont tous deux nécessaires pour maintenir l'activation microgliale et astrogliale avant même la perte neuronale manifeste, soulignant le rôle du TNF-α dans le déclenchement de la neuroinflammation induite par les cellules gliales (Barcia, 2011). Des études mécanistiques révèlent en outre que le régulateur RGS5 :
- Fait passer la signalisation du TNFR2 de protectrice à pro-inflammatoire dans les astrocytes.
- Amplifie l'activation du TNFR1
- Renforce l'agrégation de l'alpha-synucléine, la neurodégénérescence et la mortalité dans les modèles de la maladie de Parkinson (Yin, 2023).
La conversion des astrocytes en un état A1 neurotoxique représente un autre mécanisme pathologique dans lequel le TNF-α est indispensable. Les microglies libèrent de l'IL-1α, du TNF-α et du C1q, qui ensemble convertissent les astrocytes en ce phénotype nocif. Des astrocytes A1 ont été identifiés dans le cerveau post-mortem de patients atteints de la maladie de Parkinson (Liddelow, 2017). Dans les modèles animaux de la MP (modèle α-syn PFF de la MP sporadique et modèle de souris hA53T Tg), l'alpha-synucléine pathologique favorise également la formation d'astrocytes A1, et le blocage de ce processus protège les neurones dopaminergiques et préserve la fonction motrice (Yun, 2018). Ces astrocytes A1:
- Perdent leur capacité à fournir un soutien trophique et synaptique.
- Sécrètent des facteurs neurotoxiques, notamment des composants du complément et des ROS.
- Exacerbent les lésions neuronales (Liddelow, 2017).
Des études chez l'homme confirment le rôle pathogène du TNF-α astrocytaire dans la MP. Le séquençage d'ARN à noyau unique du mésencéphale humain atteint de MP révèle une activation pan-gliale avec un phénotype astrocytaireCD44high et des trajectoires de signalisation des cytokines, ce qui correspond à un tonus inflammatoire chronique dans la niche nigrale (Smajić, 2022). Dans les astrocytes humains en culture, le TNF-α et les fibrilles d'α-synucléine déclenchent des états immunitaires réactifs avec une respiration mitochondriale altérée, mettant en évidence des faiblesses métaboliques sensibles au TNF qui correspondent à la pathogenèse de la MP (Russ, 2021). Cette cascade pathogène est encore amplifiée par le complément C4, qui :
- Renforce les réponses pro-inflammatoires astrocytaires à l'alpha-synucléine.
- Favorise l'apoptose neuronale et la pathologie synaptique (Zou, 2025).
De plus, les astrocytes humains sensibilisés à l'inflammation altèrent la morphogenèse microvasculaire et perturbent l'intégrité de la BHE dans les modèles de puces cérébrales (de Rus Jacquet, 2023). Sur le plan mécanique, le TNF conduit les astrocytes dans un état de type phase aiguë dépendant de STAT3 qui transmet l'inflammation systémique à l'endothélium cérébral, favorisant ainsi la rupture de la BHE dans la MP (Kim, 2022).
Dans l'ensemble, ces résultats positionnent le TNF-αastrocytaire comme un mécanisme central qui relie la pathologie de l'α-synucléine, l'amplification gliale, le dysfonctionnement synaptique et l'altération vasculaire dans la MP.
Sclérose latérale amyotrophique (SLA)
La signalisation du TNF-α astrocytaire est apparue comme un facteur précoce et persistant de vulnérabilité des motoneurones dans la SLA. Des niveaux élevés de TNF-α sont systématiquement détectés dans le LCR et le sang des patients atteints de SLA, en corrélation avec la progression de la maladie (Jiang, 2022). De même, chez les souris SOD1G93A, le TNF-α et ses récepteurs sont régulés à la hausse dans la moelle épinière avant même l'apparition des symptômes, ce qui suggère que cette voie contribue à la pathogenèse précoce (Brambilla, 2016). Dans les co-cultures d'astrocytes et de motoneurones dérivées de ces modèles, le TNF-α lié à la membrane augmente dans les motoneurones tandis que les niveaux de TNFR2 diminuent, créant un déséquilibre dans la signalisation des récepteurs. Il convient de noter que:
- La suppression du TNFR2, mais pas du TNFR1, sauve la survie des motoneurones, identifiant l'axe mTNF-αTNFR2 comme un médiateur clé de la neurotoxicité (Tortarolo, 2015).
- À l'inverse, la signalisation TNFR1 peut exercer un effet protecteur, peut-être par la libération astrocytaire du facteur neurotrophique dérivé des glies (GDNF), car son ablation aggrave les résultats (Brambilla, 2016).
Ces résultats soulignent que les contributions du TNFR1 et du TNFR2 à la pathologie de la SLA dépendent fortement du contexte, variant entre les systèmes de co-culture isolés et les environnements in vivo complexes. En outre, le TNFR1 et le TNFR2 activent tous deux la voie ASK1/p38MAPK (apoptosis signal-regulated kinase 1), qui joue un rôle essentiel dans la mort des motoneurones. L'inhibition de la p38 MAPK protège les motoneurones dans les modèles de co-culture d'astrocytes/MN dérivés de souris SOD1G93A, soulignant le rôle de cette voie en tant qu'effecteur clé en aval de la signalisation du TNF dans la SLA (Dewil, 2007). Parallèlement:
- Dans les astrocytes porteurs du gène FUS mutant, le TNF-α active NF-κB et modifie la composition des sous-unités du récepteur AMPA en réduisant l'incorporation de GluA2.
- Ce changement augmente la perméabilité au calcium et le stress excitotoxique dans les motoneurones (Kia, 2018 ).
- La perte génétique de TNF-α ou la neutralisation pharmacologique dans les modèles FUS-SLA sauve le comportement moteur et la survie des neurones, identifiant le TNF-α dérivé des astrocytes comme le signal toxique immédiat (Jensen, 2022).
Le TNF-α astrocytaire façonne également le milieu inflammatoire, entraînant des réponses gliales et immunitaires qui aggravent la pathologie de la SLA. Les mécanismes clés comprennent :
- L'activation inductible du NF-κB dans les astrocytes favorise la prolifération microgliale et le recrutement des leucocytes périphériques, accélérant ainsi la progression des symptômes (Ouali Alami, 2018).
- La triade de signalisation microgliale IL-1α/TNF/C1q induit un phénotype astrocytaire A1 neurotoxique, et la perturbation de cet axe prolonge la survie chez les souris SOD1G93A (Guttenplan, 2020).
- Les astrocytes porteurs de mutations SOD1 adoptent un état réactif et dysfonctionnel induit par le stress mitochondrial, l'altération du protéasome et l'activation immunitaire innée, que la signalisation TNF-α exacerbe.
- Bien que le facteur de croissance des fibroblastes 4 (FGF4) puisse restaurer temporairement l'homéostasie astrocytaire, le TNF-α annule cette protection en maintenant l'activité NF-κB, laissant les motoneurones vulnérables (Velasquez, 2024).
Le TNF-α astrocytaire perturbe l'homéostasie du glutamate, renforçant encore le stress excitotoxique dans la SLA. Plus précisément, la signalisation du TNF-α via le TNFR1 astrocytaire:
- Supprime l'expression de l'EAAT2/GLT-1.
- Stimule la libération de glutamate.
- Conduit à une altération de la clairance du glutamate et à un stress excitotoxique sur les motoneurones, un mécanisme compatible avec la pathologie de la SLA humaine (Jiang, 2019).
Cette perturbation agit en synergie avec les changements induits par le TNF dans la composition des récepteurs AMPA, renforçant l'excitotoxicité induite par le calcium (Kia, 2018; Jensen, 2022).
Dans l'ensemble, le TNF-α astrocytaire agit comme un point de convergence reliant l'inflammation, le déséquilibre des récepteurs et l'excitotoxicité dans la SLA. En amplifiant simultanément les cascades inflammatoires, en modifiant le traitement du glutamate et en augmentant la perméabilité des récepteurs excitateurs, le TNF-α accélère la dégénérescence des motoneurones et favorise la progression de la maladie.
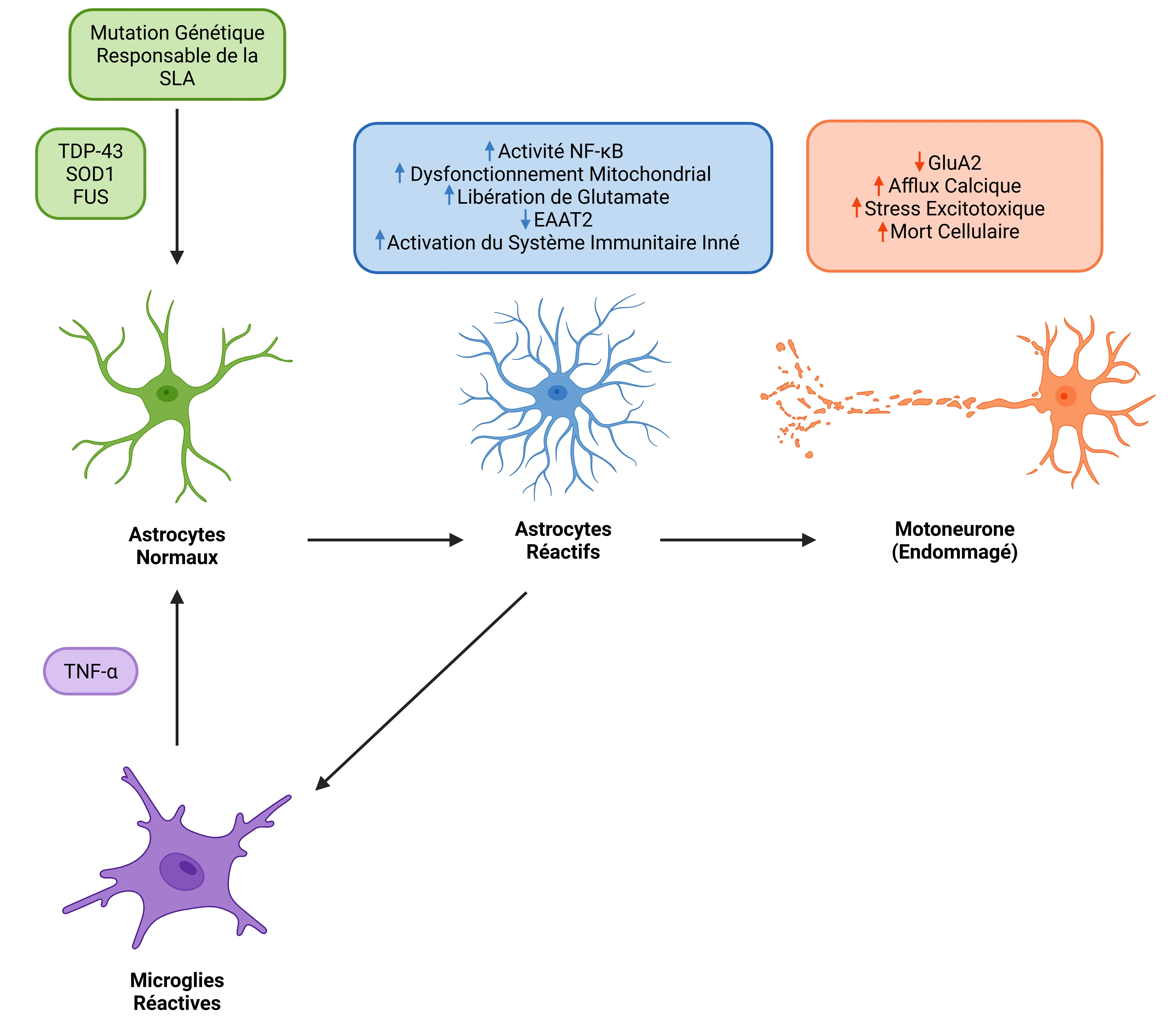
Le TNF-α en tant que facteur déterminant de la réactivité astrocytaire et de la dégénérescence des motoneurones dans la SLA. Dans la SLA associée à des mutations des gènes TDP-43, SOD ou FUS, le TNF-α libéré par les microglies réactives favorise la réactivité astrocytaire via la signalisation TNFR. Cela active NF-κB dans les astrocytes et altère la clairance du glutamate, entraînant un stress excitotoxique et la mort des motoneurones. Les astrocytes réactifs amplifient encore davantage la neuroinflammation, créant une boucle de rétroaction qui accélère la perte de motoneurones.
Sclérose en plaques (SEP)
Dans la sclérose en plaques (SEP), les astrocytes apparaissent comme des médiateurs essentiels des effets du TNF-α, reliant les cascades inflammatoires aux processus neurotoxiques et réparateurs. Le TNF-α est systématiquement élevé dans le liquide céphalo-rachidien (LCR) et détecté dans les lésions actives aiguës et chroniques de la SEP, où des niveaux plus élevés sont corrélés à une invalidité plus grave et à une progression accélérée de la maladie (Sharief, 1991; Kosa, 2022). La pathologie au niveau des lésions confirme la régulation à la hausse du TNF dans les plaques actives aiguës et chroniques, alignant la signalisation du TNF avec les sites de lésions tissulaires actives (Mazziotti, 2024).
Un mécanisme clé par lequel les astrocytes amplifient la signalisation locale du TNF implique le traitement protéolytique du TNF lié à la membrane. Au sein des lésions actives de la SEP, les astrocytes réactifs régulent à la hausse l'ADAM17/TACE, qui clive le tmTNF en sa forme soluble. Ce changement active préférentiellement le TNFR1, renforçant les cascades apoptotiques et pro-inflammatoires (Plumb, 2006). Dans l'encéphalomyélite auto-immune expérimentale (EAE), il a été démontré que la signalisation TNFR1 astrocytaire entraînait un remodelage synaptique de l'hippocampe et des troubles cognitifs, reliant directement la signalisation TNF gliale au dysfonctionnement du réseau (Habbas, 2015). Des études de sauvetage génétique de suivi ont confirmé que le rétablissement spécifique du TNFR1 dans les astrocytes suffit à rétablir le phénotype synaptique et comportemental de l'EAE (Di Castro, 2022). Ensemble, ces résultats positionnent les astrocytes non seulement comme des répondeurs, mais aussi comme des amplificateurs de la neurotoxicité médiée par le TNF.
Dans le même temps, les astrocytes sont également capables d'exploiter les voies protectrices du TNF, ce qui souligne leur double rôle dans la SEP. Ils expriment à la fois le TNFR1 et le TNFR2, et de plus en plus de preuves montrent que la signalisation du TNFR2 favorise la neuroprotection et la réparation. Dans les modèles de démyélinisation, l'activation du TNFR2 astrocytaire favorise les phénotypes anti-inflammatoires et soutient la remyélinisation par des mécanismes tels que le recrutement d'oligodendrocytes induit par le pCXCL12 (Brambilla, 2011; Patel, 2012). Des études récentes ajoutent que le TNFR2 limite les programmes astrocytaires pro-inflammatoires et renforce le soutien trophique à la myélinisation, le mettant en évidence comme un contrepoids aux dommages médiés par le TNFR1 (Raphael, 2019; Pegoretti, 2023).
Ces connaissances mécanistiques aident à expliquer pourquoi les tentatives cliniques de thérapie anti-TNF à large spectre dans la SEP ont échoué:
- Le blocage non sélectif du TNF a été associé à
- Une exacerbation paradoxale des événements démyélinisants
- Des syndromes inflammatoires de novo du SNC (W. Xie, 2024)
Au lieu de cela, l'orientation thérapeutique s'est orientée vers la modulation sélective de la signalisation du TNF (Brambilla, 2011; Pegoretti, 2023):
- Les stratégies prometteuses dans les études précliniques et translationnelles comprennent
- Inhibition du TNFR1
- Neutralisation du sTNF
- Épargner ou agoniser le TNFR2
Dans l'ensemble, ces résultats positionnent le TNF comme une cytokine essentielle qui orchestre les réponses astrocytaires dans la SEP, faisant constamment pencher la balance entre lésion et réparation. La double nature de la signalisation astrocytaire du TNF met en évidence le défi et l'opportunité du ciblage thérapeutique, faisant de la modulation sélective des récepteurs une stratégie intéressante pour la neuroprotection et la remyélinisation.
Quelles stratégies thérapeutiques ciblent la signalisation du TNF-α dans les astrocytes?
Les stratégies thérapeutiques actuelles peuvent être regroupées en quatre approches principales, chacune ciblant un aspect distinct de la signalisation du TNF-α dans les astrocytes:
Inhibiteurs des récepteurs/ligands du TNF-α
Les stratégies thérapeutiques visent de plus en plus à ajuster la signalisation du TNF-α dans les astrocytes en supprimant l'activité pathologique du TNFR1 tout en préservant ou en renforçant les fonctions réparatrices du TNFR2. Cet équilibre est essentiel, car l'inhibition sélective du TNFR1 ou la neutralisation du sTNF peut épargner la signalisation tmTNF-TNFR2, qui soutient les programmes trophiques et de remyélinisation au sein du SNC (Fischer, 2020).
Les principales approches comprennent:
- Antagonistes sélectifs des récepteurs
- L'atrosimab, un antagoniste monovalent spécifique du TNFR1 humain, a démontré son efficacité dans des modèles inflammatoires, notamment le modèle EAE, validant ainsi le blocage sélectif du TNFR1 comme une intervention pertinente centrée sur les astrocytes.
- D'autres études précliniques étendent ces avantages à la neurodégénérescence aiguë, soulignant le potentiel neuroprotecteur plus large de cette classe de médicaments (Ortí-Casañ, 2023).
- Stratégies ciblées sur les ligands
- Le TNF dominant négatif et le XPro1595 (pegipanermin) neutralisent le sTNF tout en préservant la signalisation mTNF-TNFR2 (MacPherson, 2017; De Sousa Rodrigues, 2019).
- L'inhibition de l'ADAM17/TACE empêche le clivage du tmTNF en sTNF, favorisant ainsi davantage les voies TNFR2 (L. Xie, 2024).
Malgré ces résultats prometteurs, la transposition clinique reste difficile, car la plupart des produits biologiques ont une faible pénétration à travers la BHE. Cette limitation a stimulé le développement de navettes moléculaires et de systèmes d'administration ciblant le SNC (Kouhi, 2021). De plus, l'expérience clinique avec des inhibiteurs systémiques non sélectifs du TNF a montré une aggravation paradoxale des conditions de démyélinisation, soulignant l'importance des stratégies sélectives des récepteurs et des ligands dans les contextes neuroinflammatoires (Mazziotti, 2024).
Cibler les voies de signalisation en aval
Une autre approche consiste à moduler les voies de signalisation en aval des récepteurs du TNF, en particulier NF-κB et MAPK, qui régulent la réactivité des astrocytes.
- Cibles clés en aval
- NF-kB: l'activation spécifique des astrocytes amplifie la neuroinflammation in vivo, ce qui suggère qu'une inhibition sélective pourrait être bénéfique, bien que de nouvelles études sur des cellules humaines montrent que ses effets sur les neurones peuvent être à la fois protecteurs et nocifs selon le contexte (Giovannoni, 2020).
- p38MAPK: les inhibiteurs sélectifs tels que le MW150 suppriment la neuroinflammation et améliorent la cognition dans les modèles de MA, ce qui suggère un potentiel de transposition (Frazier, 2024).
- MEK1/2 (voie ERK): l'inhibition rétablit la morphogenèse microvasculaire dans les astrocytes présentant une mutation associée à la maladie de Parkinson. Dans des conditions normales, la voie MEK/ERK aide les astrocytes à réguler les réponses au stress et à mettre en place une signalisation inflammatoire équilibrée. Cependant, dans le cas d'une maladie, ce rôle protecteur est amplifié de manière pathologique, ce qui fait de la voie MEK/ERK un levier maniable pour la stabilisation de la BHE et une cible thérapeutique prometteuse dans la neurodégénérescence (de Rus Jacquet, 2023).
- JNK: l'inhibition limite la libération de CXCL1 par les astrocytes et le stress neuronal en aval, positionnant JNK comme une cible de voie parallèle (Zhang, 2022).
Modulation des phénotypes astrocytaires
Une stratégie thérapeutique complémentaire vise à reprogrammer les astrocytes pour les éloigner des phénotypes neurotoxiques et les orienter vers des états homéostatiques ou réparateurs. Le blocage de l'axe IL-1α/TNF/C1q empêche la conversion en astrocytes de type A1 et préserve la viabilité des neurones et des oligodendrocytes in vivo, fournissant l'une des démonstrations les plus convaincantes que la modulation du phénotype des astrocytes est thérapeutique (Liddelow, 2017). D'autres mécanismes prometteurs incluent :
- L'inhibition de l'ELOVL1, qui bloque la libération de particules lipidiques toxiques, mettant en évidence le métabolisme lipidique comme une autre vulnérabilité pouvant être exploitée par des médicaments (Guttenplan, 2021).
- Les inhibiteurs HDAC3, qui réduisent l'activation des gènes inflammatoires tout en préservant les fonctions de soutien des astrocytes (Clayton, 2024).
- Les agonistes des récepteurs GLP-1 (g. NLY01), qui suppriment la toxicité des astrocytes induite par les microglies et font actuellement l'objet d'essais cliniques dans le cadre de la MA et de la MP (Yun, 2018; Park, 2021).
Thérapie génique et nouveaux produits biologiques
Les thérapies géniques et biologiques élargissent encore les options de contrôle spécifique des astrocytes sur la signalisation du TNF :
- L'administration du gène IL-2 aux astrocytes augmente les cellules T régulatrices locales et réduit la neuroinflammation sans affecter l'immunité systémique (Yshii, 2022).
- Les progrès réalisés dans le domaine des vecteurs AAV utilisant des promoteurs GFAP, des éléments spécifiques aux astrocytes de nouvelle génération et des amplificateurs de sérotypes larges, rendent désormais possible le transfert de gènes sélectif aux astrocytes chez les rongeurs et les espèces de plus grande taille (O'Carroll, 2021; Heffernan, 2022; Gleichman, 2023).
- Les stratégies combinées, telles que l'agonisme séquentiel du TNFR2 suivi de l'antagonisme du TNFR1, améliorent les résultats dans les modèles EAE humanisés et démontrent que le biais temporel de l'activité des récepteurs peut maximiser l'efficacité thérapeutique tout en minimisant les risques (Pegoretti, 2023).
Dans l'ensemble, le ciblage de la signalisation TNF-α astrocytaire souligne la nécessité de supprimer ses effets inflammatoires pathologiques tout en préservant les fonctions protectrices qui favorisent la réparation et la stabilité de la BHE. La principale difficulté reste la pénétration limitée des traitements actuels dans le cerveau, mais les progrès réalisés dans le domaine des inhibiteurs sélectifs, des systèmes d'administration et des interventions axées sur les astrocytes ouvrent la perspective de transformer la modulation du TNF-α en un traitement neuroprotecteur viable.
Comment le TNF-α se distingue-t-il des autres cytokines dans le processus de neurodégénérescence?
Plusieurs distinctions clés différencient le TNF-α des autres cytokines dans la neurodégénérescence, façonnant à la fois son rôle dans la progression de la maladie et son potentiel en tant que cible thérapeutique.
Fonctions distinctives du TNF-α (apoptose, nécroptose, inflammation)
Le TNF-α occupe une place unique parmi les cytokines neuroinflammatoires, car son récepteur relie la signalisation inflammatoire à la mort cellulaire programmée. Contrairement à d'autres médiateurs, le TNF-α associe l'induction génique à l'apoptose et à la nécroptose par le biais du TNFR1, engageant les voies RIPK1/RIPK3/MLKL en plus des réponses de survie et pro-inflammatoires induites par NF-κB (Holbrook, 2019; van Loo, 2023).
En revanche:
- L'IL-6 transmet ses signaux par
- la signalisation « classique » via l'IL-6R membranaire, qui favorise les réponses protectrices et régénératrices.
- la « trans-signalisation » via l'IL-6R soluble, qui amplifie l'inflammation.
- Les récepteurs IL-6 solubles agissent principalement comme des agonistes, contrairement au TNF, où les récepteurs solubles ont tendance à être antagonistes (Rose-John, 2021).
- Active les réponses biaisées STAT3, amplifie la réactivité des astrocytes et perturbe la stabilité vasculaire, contribuant à la rupture de la BHE (Mora, 2024).
- L'interféron-gamma (IFN-gamma) reprogramme les astrocytes vers la présentation d'antigènes, produisant des astrocytes réactifs sensibles à l'interféron (IRRA) au lieu d'états pro-mort induits par le TNF (Rostami, 2020; Prakash, 2024; Lee, 2023).
Ces résultats suggèrent que le TNF-α établit un lien unique entre les signaux inflammatoires innés et les voies de mort cellulaire, le positionnant plus près des voies « finales communes » de la neurodégénérescence que l'IL-6 ou l'IFN-γ.
Synergie et divergence du TNF-α avec d'autres cytokines dans les astrocytes
Sur le plan fonctionnel, le TNF-α agit souvent comme une cytokine d'amorçage qui prépare les astrocytes à des phénotypes inadaptés, en particulier lorsqu'il agit conjointement avec d'autres médiateurs.
Les interactions clés comprennent :
- TNF-α + IL-1α + C1q: conduit les astrocytes à des états de type A1, entraînant une perte de la fonction de soutien des synapses et la mort des neurones et des oligodendrocytes (Liddelow, 2017).
- TNF-α + IL-1β : forte activation de la signalisation NF-κB dans les astrocytes dérivés d'iPSC humaines, remodelant leur structure et induisant un état immunitaire réactif puissant (Hyvärinen, 2019).
Ces résultats soulignent que, tandis que le TNF-α agit en synergie avec l'IL-1α (associée au C1q) et l'IL-1β pour induire des états neurotoxiques et immunoréactifs des astrocytes, des cytokines telles que l'IFN-γ et l'IL-6 activent des voies divergentes, mettant en évidence la manière dont des programmes de cytokines distincts modèlent le comportement des astrocytes dans la maladie.
Signatures spatiales et unicellulaires des astrocytes induits par le TNF
La transcriptomique unicellulaire et spatiale montre que les astrocytes sensibles au TNF-α forment des états uniques, pertinents pour la maladie :
- Dans la MA
- le séquençage d'ARN simple (snRNA-seq) montre que les astrocytes situés à proximité des plaques perdent leurs programmes génétiques de soutien normaux et activent à la place les voies inflammatoires et du complément induites par le TNF (Dai, 2023).
- La transcriptomique spatiale confirme en outre que ces astrocytes situés à la périphérie des plaques sont fortement influencés par la signalisation TNF/IL-1 et se regroupent autour des neurones en dégénérescence (He, 2024).
- Des méta-analyses montrent des augmentations parallèles du TNF-α, de l'IL-6 et de l'IL-1β tout au long du continuum de la MA, qui sont directement corrélées à l'aggravation du déclin cognitif (Lista, 2024; Serna, 2025).
- Dans la maladie de Parkinson
- le séquençage de l'ARNsn démontre une activation généralisée des cellules gliales, y compris un sous-ensemble d'astrocytes à forte expression de CD44 dont l'expression génique reflète une inflammation chronique induite par le TNF/IL-1 dans la substance noire (Smajić, 2022).
Sélectivité thérapeutique du TNF-α par rapport à d'autres cytokines
Le TNF-α se distingue sur le plan thérapeutique car son architecture de signalisation permet de cibler sélectivement les voies TNFR1 nocives tout en épargnant les fonctions protectrices du TNFR2. Ce type d'équilibre sélectif est propre au TNF et ne peut être obtenu avec le blocage de l'IL-6 ou de l'IFN-γ (Fischer, 2020; Papazian, 2021).
- Les traitements anti-TNF non sélectifs peuvent aggraver l'auto-immunité du SNC (Mazziotti, 2024).
- Les traitements par IL-6 (g. sgp130Fc) bloquent l'inflammation tout en laissant intacte la signalisation « classique » protectrice de l'IL-6, mais ne traitent pas les voies de mort cellulaire propres à la signalisation du récepteur du TNF (Rose-John, 2021).
- Les traitements par IFN-γ modulent les fonctions immunitaires des astrocytes, telles que la présentation des antigènes, plutôt que les cascades apoptotiques et nécroptotiques induites par le TNF (Prakash, 2024; Lee, 2023).
Ensemble, ces comparaisons montrent que le TNF-α est unique parmi les cytokines, car sa signalisation peut être ajustée de manière thérapeutique : le blocage sélectif du TNFR1 ou du sTNF supprime les voies neurotoxiques et de mort cellulaire tout en préservant la réparation médiée par le TNFR2, un équilibre thérapeutique que les interventions de l'IL-6 ou de l'IFN-γ ne peuvent pas atteindre.
Dans l'ensemble, les preuves montrent que le TNF-α façonne le comportement des astrocytes d'une manière plus directement liée à la progression neurodégénérative que les autres cytokines. En couplant les signaux inflammatoires aux voies de mort cellulaire, en amplifiant les états d'astrocytes inadaptés et en entraînant des changements transcriptionnels spécifiques à certaines régions, le TNF-α place les astrocytes au centre des mécanismes pathologiques et des opportunités thérapeutiques.
Notre équipe se fera un plaisir de répondre à toutes vos questions concernant le rôle du TNF-α dans les astrocytes et son implication dans les maladies neurodégénératives, ou de vous fournir des informations spécifiques sur les modèles de MA, SLA et MP que nous utilisons pour les études d'efficacité thérapeutique.
En savoir plus sur nos modèles de maladies neurodégénératives
Contenu connexe
Informations actualisées sur le TNF-α et les microglies dans les maladies neurodégénératives et meilleures pratiques liées à l'évaluation des agents thérapeutiques dans les modèles animaux de maladies neurodégénératives.
TNF-α et microglie dans les maladies neurodégénératives
Un aperçu de la fonction du facteur de nécrose tumorale alpha (TNF-α) dans la microglie et de sa contribution à la progression de la neurodégénérescence.
Qu'est-ce que l'IL-1β?
Présentation générale de l'IL-1β, de son rôle pro-inflammatoire dans les maladies systémiques et neurologiques, et des stratégies thérapeutiques impliquant l'antagonisme de l'IL-1β.
Dysfonctionnement mitochondrial dans les microglies et les astrocytes
Le rôle du dysfonctionnement mitochondrial dans les microglies et les astrocytes dans les maladies neurodégénératives, notamment la maladie d'Alzheimer, la maladie de Parkinson et la SLA.
Microglie, astrocytes et α-synucléine dans la maladie de Parkinson
Comment l'α-synucléine influence les microglies et les astrocytes dans la maladie de Parkinson et d'autres synucléinopathies.
Interleukine-1 bêta (IL-1β) et maladies neurodégénératives
Le rôle de l'IL-1bêta dans les maladies neurodégénératives, notamment la maladie d'Alzheimer (MA), la maladie de Parkinson (MP) et la sclérose latérale amyotrophique (SLA).
Microglie, astrocytes et protéine tau dans les maladies neurodégénératives
Comment la neuroinflammation induite par les cellules gliales favorise l'agrégation et la propagation de la protéine tau ainsi que la perte neuronale dans la maladie d'Alzheimer et d'autres tauopathies.