Amyotrophic Lateral Sclerosis (ALS) Mouse Models
Global preclinical neuroscience CRO with unmatched expertise in ALS mice & translational biomarkers.
Validated ALS models (TDP-43 mice) for therapeutic efficacy, MoA, and target engagement studies.
Biospective’s validated ALS animal models (including rNLS8 TDP-43ΔNLS mice and our proprietary "Low Dox" model) are purpose-built for neuroscience drug development. Our ALS mice feature motor deficits, cytoplasmic mislocalization & phosphorylated TDP-43 aggregates, neuroinflammation, and neurodegeneration. As a leading global neuroscience CRO, Biospective delivers decision-ready in vivo data supported by translational biomarkers, including neurofilament light (NfL), to biotech & pharmaceutical companies.
Biospective specializes in models of Amyotrophic Lateral Sclerosis (ALS). We have developed and characterized industry-leading ALS mouse models and have deep expertise in TDP-43 pathology. As a global preclinical neuroscience contract research organization (CRO), we support biotech & pharmaceutical drug development programs using validated ALS animal models for therapeutic efficacy, target engagement, mechanism-of-action, biodistribution, and PK/PD studies across small molecules, antisense oligonucleotides (ASOs), gene therapies, antibodies, and other biologics.
Biospective’s TDP-43 mouse models (including the conventional "Off Dox" rNLS8 model and our proprietary, slower progressing "Low Dox" TDP-43ΔNLS model) recapitulate key features of human ALS, including phosphorylated protein aggregation in brain and spinal cord neurons, axonal degeneration, neuroinflammation, muscle atrophy, denervation of the neuromuscular junction (NMJ), and motor dysfunction. Studies include translational biomarkers, such as neurofilament light chain (NfL) in plasma and CSF, advanced neuroimaging, and quantitative multiplex immunofluorescence. With fully integrated, end-to-end preclinical services, and unmatched experience conducting ALS contract research studies in animal models, Biospective enables translational Amyotrophic Lateral Sclerosis research from study design through data analysis.
Why Choose Biospective as Your ALS Models CRO?
Biospective is a neuroscience CRO with a focus on ALS animal models, strong scientific expertise, and extensive experience conducting preclinical studies in TDP-43 models.
-
Specialized ALS Mouse Models CRO: Focused exclusively on ALS and neurodegenerative disease models, not a generalist animal provider.
-
Multiple Validated ALS Models: "On Dox" and "Low Dox" rNLS8 mice are readily available for studies.
-
TDP-43 Expertise: Deep scientific expertise in TDP-43 biology and pathology, a central misfolded protein in sporadic and familial ALS.
-
Integrated Services: Fully integrated preclinical services from study design to data interpretation, ensuring seamless execution.
-
Proven Efficacy Data: Industry-standard TDP-43 efficacy datasets and extensive historical controls for robust benchmarking.
-
In-House Transgenic Mouse Colony: Large internal colony of rNLS8 (TDP-43ΔNLS) transgenic mice, enabling large scale studies with well-characterized animals.
-
Accelerated Timelines: Rapid study initiation and efficient workflows to compress timelines without sacrificing quality.
-
Translational Biomarkers: Advanced biomarkers (NfL CSF/blood assays, in vivo MRI) that bridge preclinical findings to clinical outcomes.
- Flexible Study Designs: Our scientists work with your team to customize the study design to best fit your goals.
-
Global Support: Experience supporting biotech and pharmaceutical industry clients worldwide with responsive project management and communication.
Our scientists work as an extension of your internal team, collaborating closely to ensure scientific rigor, reproducibility, and translational relevance at every stage of your ALS drug discovery & development programs.
TDP-43 Mouse Models – Our Core Expertise
Biospective specializes in disease-relevant TDP-43 mouse models for ALS drug development.
TDP-43 protein aggregation is central to Amyotrophic Lateral Sclerosis pathophysiology. Biospective has built specialized capabilities around TDP43–based ALS animal models, making this competency a core differentiator of our CRO services.
These mouse models enable direct evaluation of target engagement and downstream neurodegenerative processes under pathological conditions. Notably, our TDP-43 models recapitulate key features of human ALS – including pTDP-43 (p409/410) aggregates, neurodegeneration, neuroinflammation, motor impairments, muscle weakness and wasting, and altered EMG. Our animal model portfolio emphasizes reproducibility, well-defined phenotypes, and the integration of behavioral, imaging, biochemical, molecular, and histopathological endpoints to enable comprehensive in vivo ALS therapeutic efficacy and exploration of mechanism-of-action studies.
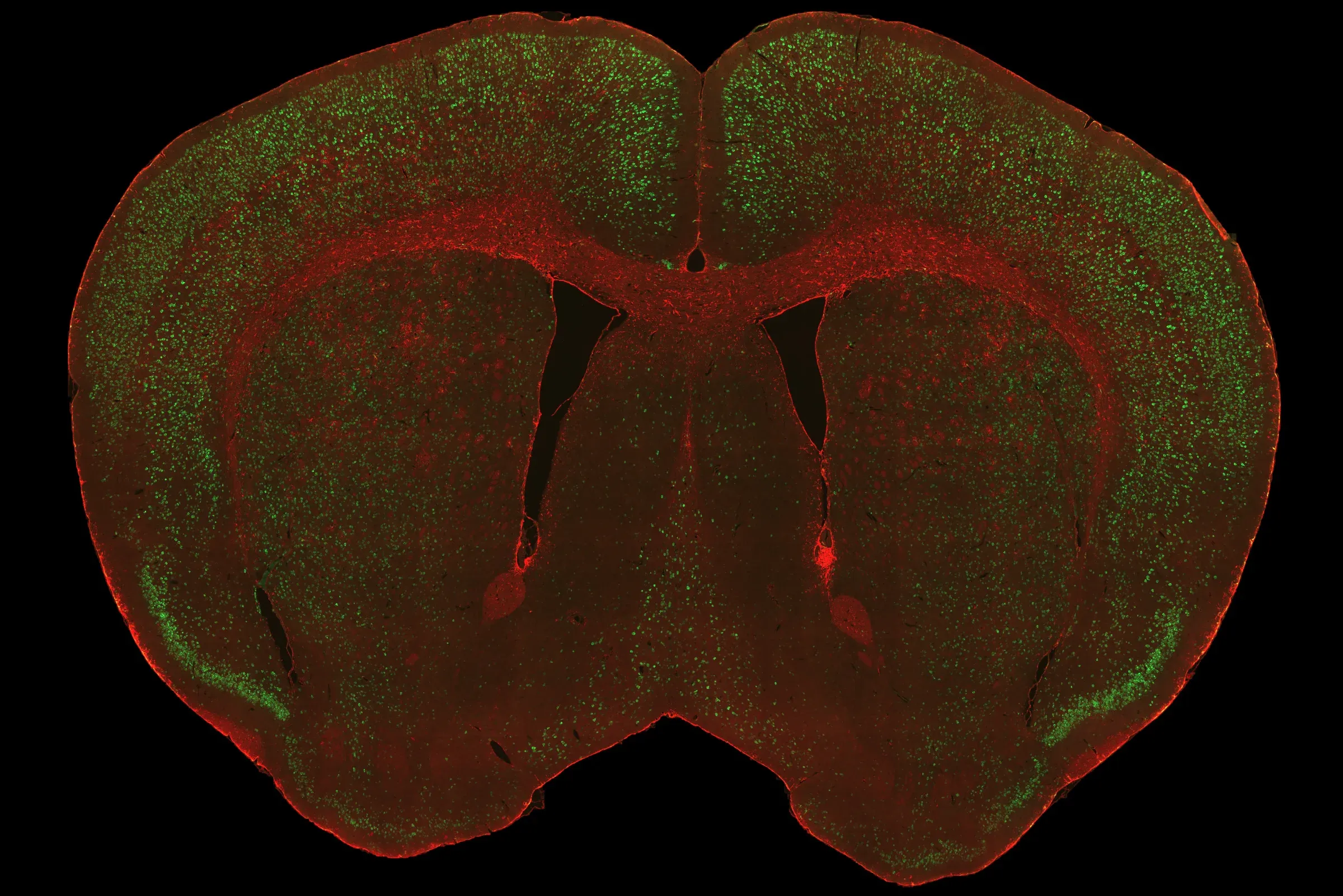
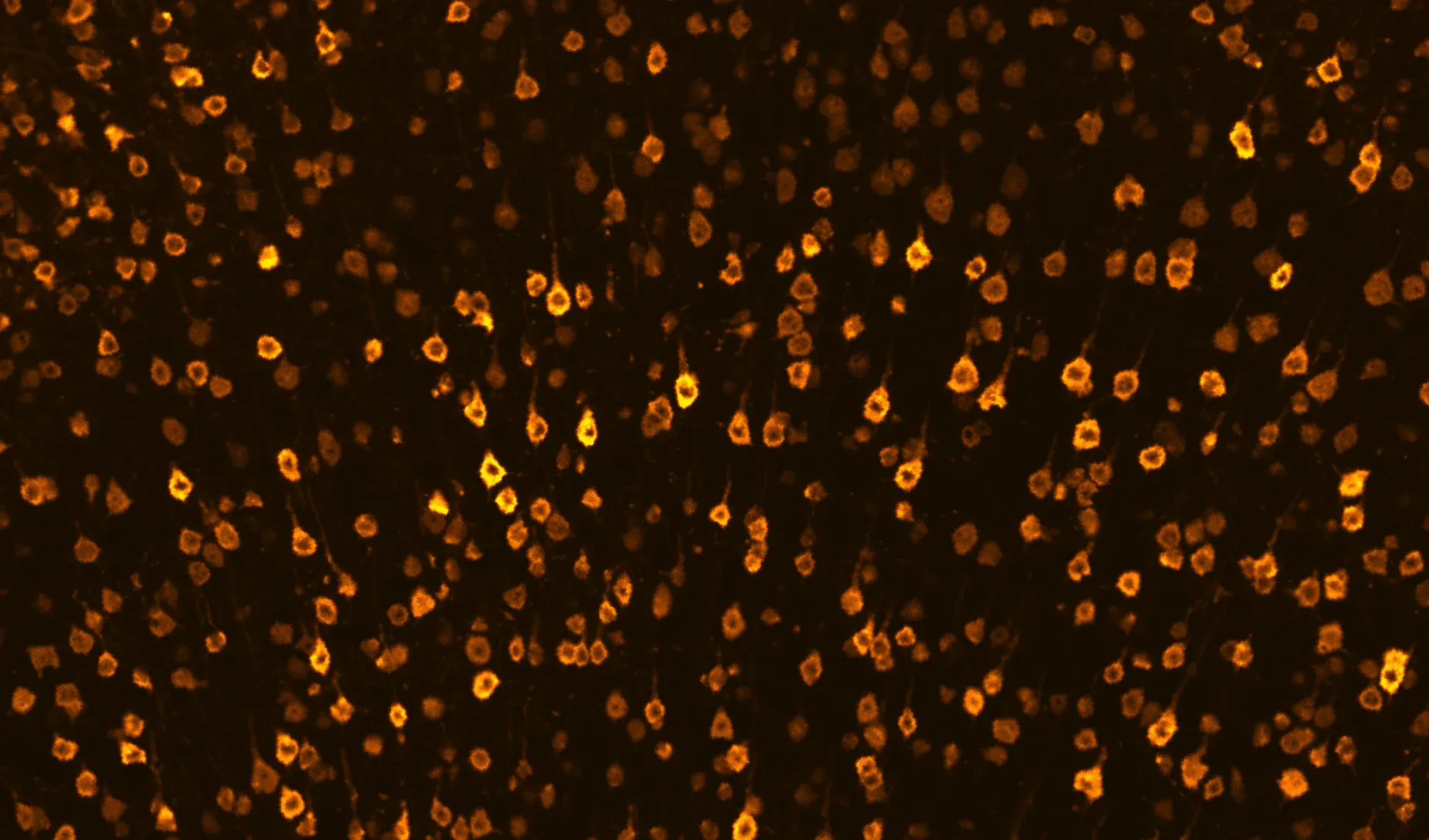
Representative visualizations of human TDP‑43 pathology in the rNLS8 mouse model, showing a coronal brain sections with TDP‑43 and astrocytes (top), TDP‑43 in cerebral cortex (middle), and mislocalization of hTDP‑43 in the cytoplasm of neurons in the motor cortex (bottom).
TDP-43ΔNLS (rNLS8) Model
The cytoplasmic mislocalization of misfolded TDP-43 aggregates that characterizes human ALS can be recapitulated in the mouse brain. In this inducible transgenic mouse model, human wild-type TDP-43 with a defective nuclear localization signal (NLS) is expressed under the NEFH promoter. This model is considered one of the best ALS animal models for drug testing given its strong translational relevance.
Disease Features Modeled:
- Cytoplasmic phosphorylated TDP-43 pathology in brain and spinal cord
- Neurodegeneration (loss of vulnerable neuronal populations & axonal degeneration)
- Neuroinflammation (microglial and astroglial activation)
- Muscle atrophy, muscle weakness, and NMJ denervation
- Reduced muscle CMAP amplitude and increased latency
- Measurable motor impairments
Our Low Dox TDP-43ΔNLS model is highly reproducible and widely regarded as a gold standard for testing disease-modifying therapeutics. Biospective has a decade of experience executing preclinical studies with ALS models to evaluate biodistribution, target engagement, mechanism of action, and therapeutic efficacy.
Translational Pathology and Biomarkers in ALS Models
Biospective has established a broad range of clinically-relevant disease markers to facilitate translation to clinical studies.
As a Preclinical Neuroscience CRO, we design our ALS rodent models with translational relevance to mirror key aspects of the human disease. A major differentiator of Biospective is our focus on translational biomarkers that align preclinical findings with clinical outcomes – including fluid biomarkers & advanced neuroimaging biomarkers. We incorporate:
-
TDP43–related biomarkers (pathology and spread)
-
Neuroinflammation markers (microglial/astrocyte activation)
-
Neurodegeneration endpoints (axonal degeneration, atrophy)
-
Mechanism-of-action confirmation (target/pathway engagement)
Our modeling and biomarker strategies ensure that preclinical successes meaningfully predict clinical potential, thereby de-risking the transition from animal studies to human trials.
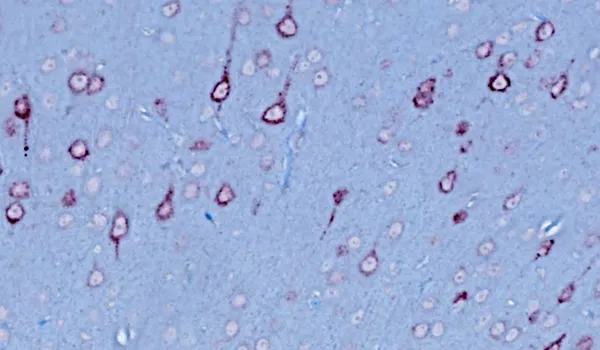
Immunohistochemistry (IHC) staining & brightfield imaging of human TDP-43 mislocalized to the cytoplasm.
TDP-43 Aggregates
Aggregates of misfolded proteins, such as TDP-43 and SOD1, are neuropathologic hallmarks of sporadic and familial forms of ALS. In >97% of cases of ALS, aggregates of TDP-43 are found mislocalized to the cytoplasm of neurons in the brain and spinal cord (Arnold, 2023). Phosphorylated aggregates are also typically observed. These characteristic features are readily observed in our TDP-43ΔNLS mouse models.
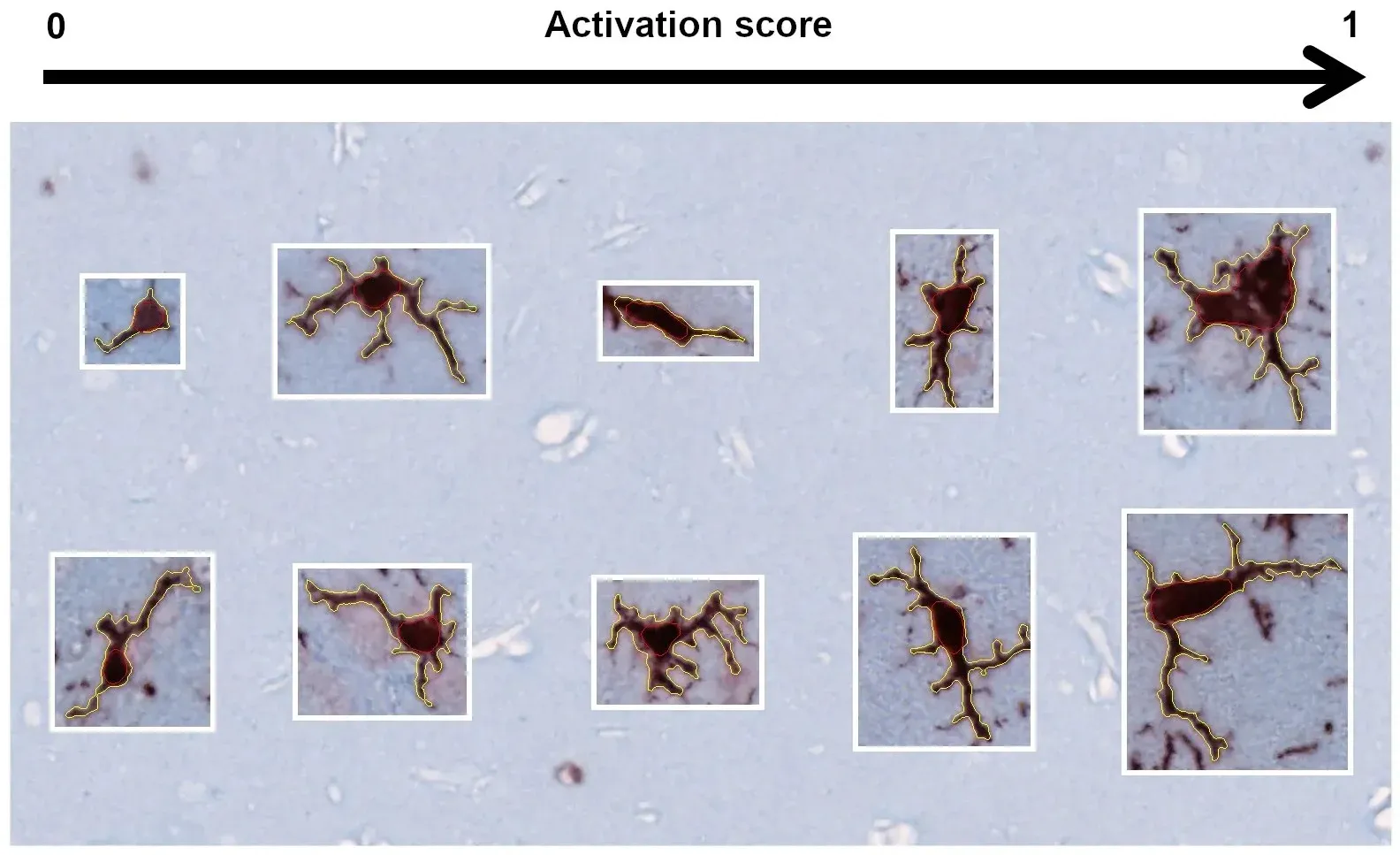
Activation scoring of microglia based on morphological features.
Activated Microglial & Reactive Astrocytes
Activated microglia and reactive astrocytes are prominent neuroinflammatory features in brains and spinal cords from ALS patients, and are thought to play a pivotal role in the disease pathogenesis (Clarke and Patani, 2020; Yang, 2024). We observe time-dependent increases in neuroinflammation in our TDP-43 mouse model. In addition to increased Iba-1 and GFAP immunoreactivity, we have found a strong relationship between activation and motor phenotype using algorithms that we developed to measure microglia and astrocyte morphology.
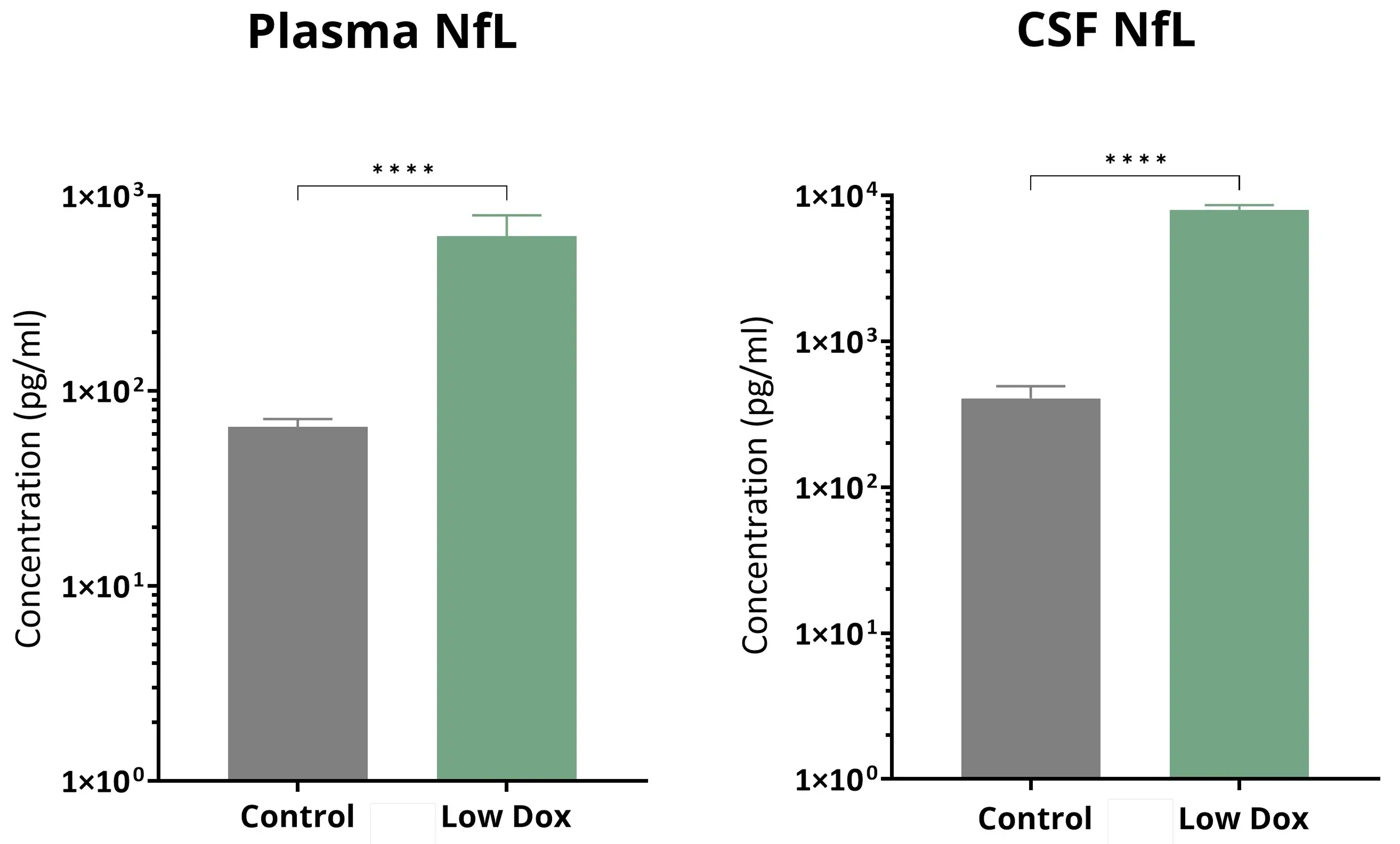
Plasma and CSF levels of neurofilament light chain (NfL) from control and Low Dox rNLS8 mice.
Elevated Neurofilament Light in Plasma & CSF
Neurofilament light chain (NfL) is increased in the plasma and CSF of ALS patients (Benatar, 2023). Neurofilament light measurements are routinely used in ALS clinical trials. The accelerated approval of tofersen (Qalsody) supported by reduction in neurofilament light levels indicated the FDA’s acceptance of this measures as disease biomarker. We observe highly significant increases in plasma & CSF levels of neurofilament light in our TDP-43 mouse models. Young et al. have demonstrated the ability of a small molecule PIKfyve inhibitor, AIT-101 (INN: apilimod, aka LAM-002A), to decrease neurofilament light levels in our “Low Dox” TDP-43ΔNLS model.
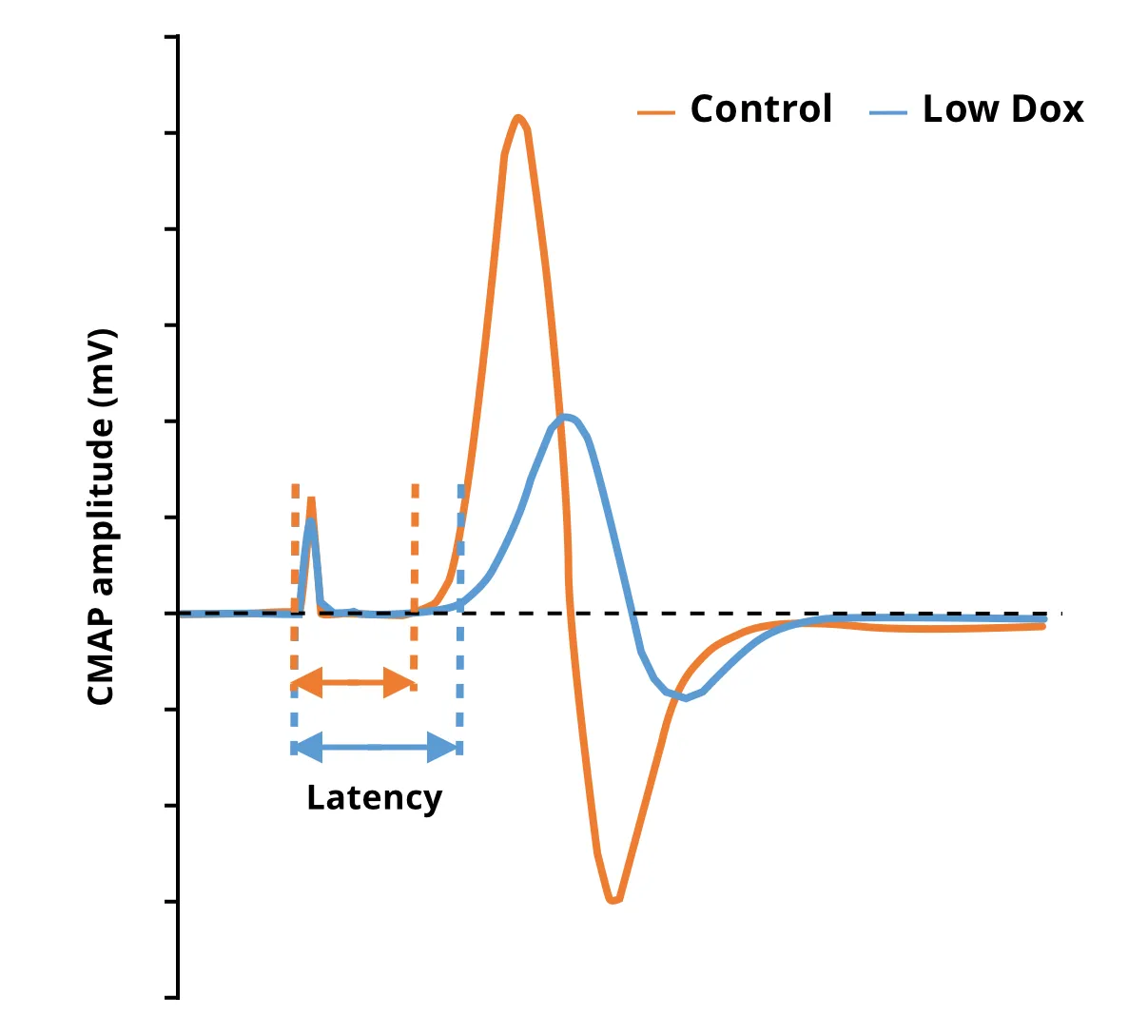
Compound Muscle Action Potential (CMAP) measures in Low Dox rNLS8 mice show significantly reduced amplitude and increased latency compared to control mice.
NMJ Denervation & Morphological Alterations
Electrophysiology (e.g. electromyography [EMG]) is a standard test used in ALS patients for diagnosis and monitoring of disease. Altered EMG measures, such as the Compound Muscle Action Potential (CMAP) reflect neuromuscular denervation due to the loss of spinal motor neurons (Sleutjes, 2021). Denervation and morphological changes of the neuromuscular junction (NMJ) have also been found in muscles from ALS patients (Bruneteau, 2015). In our “Low Dox” TDP-43ΔNLS model, we have found significantly reduced CMAP amplitude and increased latency compared to control mice. Using multiplex immunofluorescence, we have also identified NMJ alterations consistent with denervation.
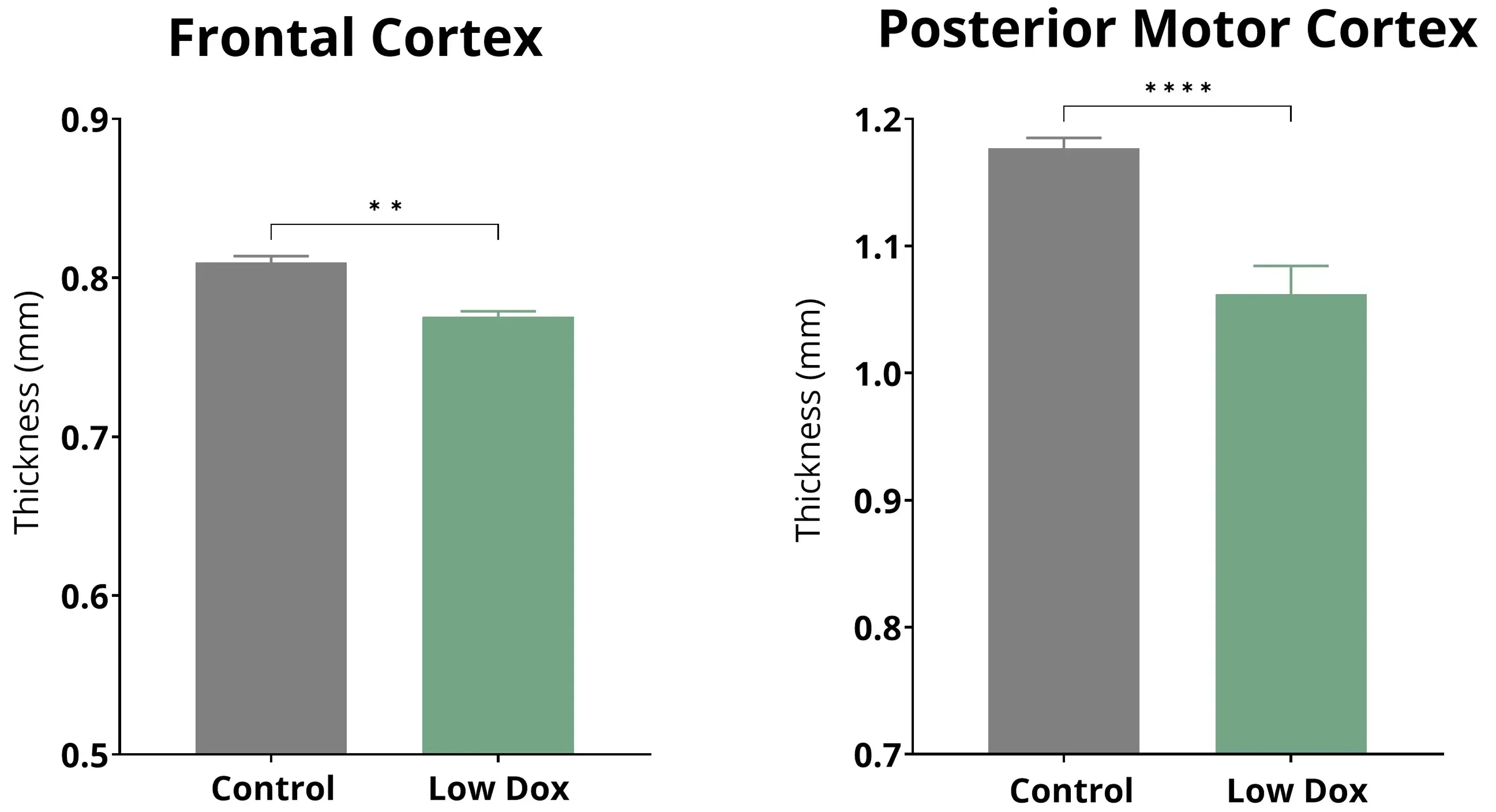
Quantitative in vivo MRI measures of cortical thickness in Low Dox rNLS8 mice shows cortical thinning compared to control animals.
Regional Brain Atrophy
Neuroimaging biomarkers are widely used in clinical trials of neurodegenerative diseases, including ALS. MRI-derived regional volume and cortical thickness measures are highly sensitive to brain atrophy and allow for monitoring disease progression over time. Cortical thinning has been shown in motor and non-motor brain regions in ALS (Yang, 2025). Using high-resolution, whole brain MRI acquisition and fully-automated image processing & analysis, we have shown reproducible brain atrophy (particularly in the motor and frontal cortex) in our Low Dox TDP-43ΔNLS mouse model, thereby serving as a robust in-life measure of neurodegeneration that complements other measures, such as fluid-based neurofilament light chain measures.
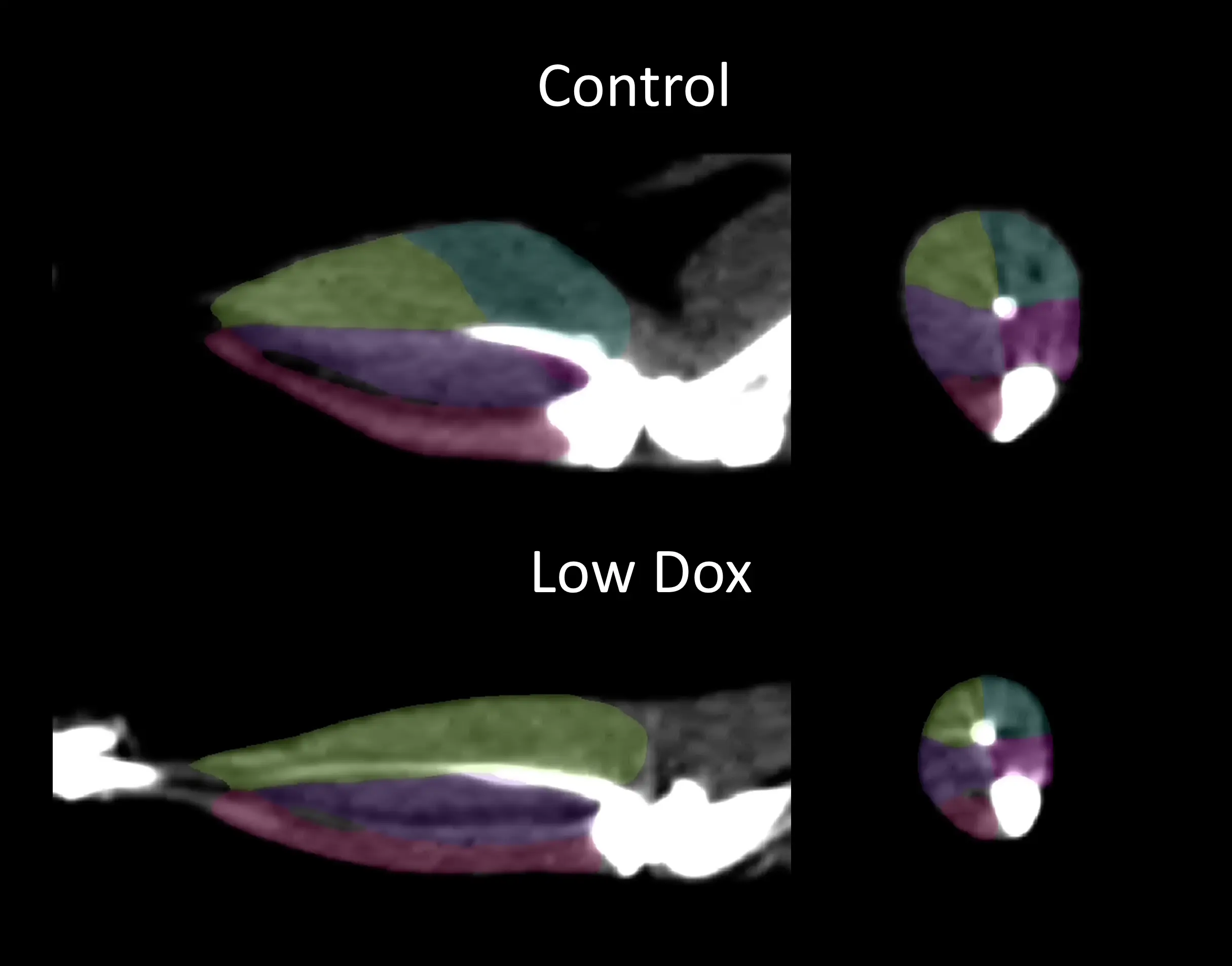
Longitudinal in vivo CT imaging can be effectively used to measure limb muscle atrophy in TDP-43 mouse models of ALS.
Muscle Wasting & Weakness
Skeletal muscle atrophy and weakness are central clinical features of ALS (Shefner, 2023). Non-invasive imaging techniques have been used to quantify muscle atrophy in ALS patients (Jenkins, 2013; Jenkins, 2018; Wilcox, 2021; Klickovic, 2024). We have used microCT to perform longitudinal measurements of hindlimb muscle loss in our Low Dox TDP-43ΔNLS mouse model and have found highly significant differences compared to control mice. This muscle wasting is accompanied by loss of muscle strength measured using a grip strength meter.
How are ALS Mouse Models Used in Drug Development?
We work closely with our biotech and pharmaceutical sponsors to:
-
Evaluate therapeutic efficacy and dose-response in ALS models
-
Assess target engagement and disease-modifying effects
-
Support translational biomarker strategies, including fluid biomarkers (e.g. NfL) for clinical readiness
Our ALS mouse models are optimized for in vivo testing of multiple therapeutic modalities, including both traditional and advanced approaches:
Small Molecules
-
Brain & spinal cord distribution
-
Effect on motor dysfunction
-
Reduction of pathological hallmarks (TDP-43 aggregates, NMU denervation)
RNA-Targeted Therapies
- Target knockdown verification (e.g. mRNA or protein level reduction)
-
CNS biodistribution of ASOs/siRNA
-
Translational biomarker readouts to confirm pathway engagement
Gene Therapy & Viral Vectors
-
Transgene expression levels in target regions
-
Regional biodistribution of viral vectors (e.g. AAV spread)
-
Functional rescue or disease modification outcomes (behavioral and pathological improvements)
Antibodies & Biologics
-
CNS exposure and penetration of biologics (e.g. BBB transport)
-
Human TDP-43 aggregation clearance or reduction
-
Mechanism-of-action validation (target binding, downstream signaling changes)
End-to-End ALS Preclinical CRO Services
Biospective offers fully integrated preclinical ALS contract research services.
-
Study design & model selection – expert guidance on choosing the most appropriate ALS model and designing robust studies
-
In vivo efficacy studies – execution of treatment studies with thorough monitoring of outcomes
-
Biodistribution & PK/PD – analysis of drug distribution and pharmacokinetics/pharmacodynamics in CNS and periphery
-
Target engagement assays – confirmation that the therapeutic hits its molecular target (e.g. TDP-43 reduction, pathway modulation)
-
Behavioral analysis – motor function testing (grip strength, hindlimb clasping, grill agility, etc.)
-
In vivo multi-modality imaging – MRI, PET, SPECT, DEXA, fluorescence, and bioluminescence imaging to track disease and treatment effects
-
Immunoassays – biomarker quantification in CSF, blood, and tissue (e.g. NfL, cytokines, chemokines)
-
Immunohistochemistry (IHC) & multiplex immunofluorescence (mIF) – post-mortem tissue staining & quantitative image analysis to assess pathology and therapeutic impact
-
Data analysis & reporting – rigorous quantitative analysis, statistics, and comprehensive reporting by our scientists
This end-to-end approach minimizes handoffs, accelerates timelines, and reduces risk for our sponsors by keeping all aspects of the study under one expert team.
Learn more about our characterization of these ALS mouse models, our validated measures, and our Preclinical Neuroscience CRO services.
Related Content
Up-to-date information on ALS and best practices related to the evaluation of therapeutic agents in ALS animal models.
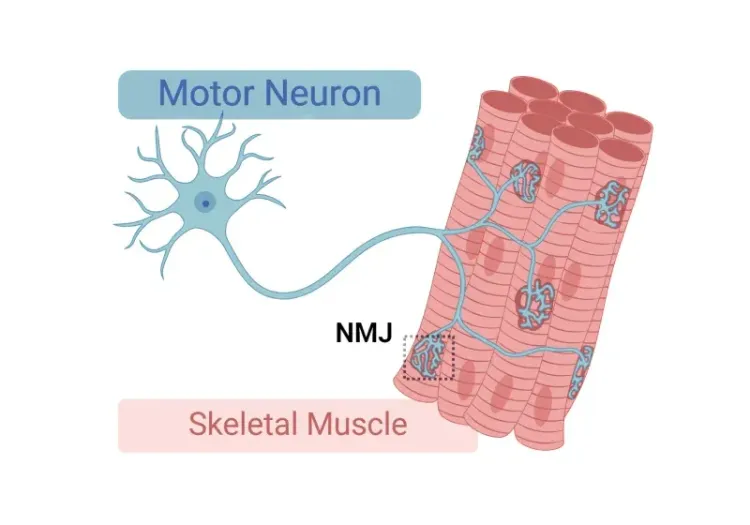
Neuromuscular Junction (NMJ) Morphology & ALS Models
Insights into neuromuscular junction (NMJ), its role in amyotrophic lateral sclerosis (ALS), and tools & methods used to study morphological changes in NMJs.
ALS Mouse Models & Spinal Motor Neurons
An overview of the involvement of spinal motor neurons in disease progression in mouse models of Amyotrophic Lateral Sclerosis (ALS).
A Guide to ALS Models for Drug Discovery
A Resource for the most effective use of research animal models (mouse & rat models) of Amyotrophic Lateral Sclerosis (ALS) for preclinical testing of therapeutics.
TDP-43 ΔNLS (rNLS8) Mice for ALS Drug Development
This resource provides information about the use of the ΔNLS (deltaNLS, hTDP-43ΔNLS, hTDP-43DeltaNLS, dNLS, TDP43 NLS, rNLS8) TDP-43 transgenic mouse model of ALS for preclinical therapeutic studies.
Microglia Morphology in ALS, Alzheimer's Disease & Parkinson's Disease
An overview of microglial morphological analysis and the applications to neurodegenerative disease research and drug discovery & development.
TDP-43: Role in ALS and Frontotemporal Dementia (FTD)
An overview of TDP-43, its physiological role, significance in ALS and FTD pathology, and therapeutic strategies involving TDP-43.
Autophagy & Neurodegenerative Diseases
An overview of how cellular autophagy plays a role in brain health and neurodegeneration.
NLRP3 Inflammasome and Neurodegenerative Diseases
An overview of the NLRP3 inflammasome and its role in neurodegenerative diseases, including Alzheimer's disease, Parkinson’s disease, and ALS.
TNF-α (TNF-alpha) & Microglia in Neurodegenerative Diseases
An overview of the function of tumor necrosis factor-alpha (TNF-α) in microglia and its contribution to the progression of neurodegeneration.
Impaired Microglia Autophagy in Neurodegenerative Diseases
How impaired microglia autophagy contributes to the progression of neurodegenerative diseases.
Microglia-Neuron Interactions & Neurodegenerative Diseases
A concise review of the direct interactions between microglia & neurons, and how these cell-to-cell interactions may be affected in neurodegenerative diseases.
Microglial Senescence and Neurodegenerative Diseases
An overview of microglial senescence and its role in neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD).
TNF-α & (TNF-alpha) Astrocytes in Neurodegenerative Diseases
An overview of TNF-α signaling in astrocytes, its role in neurodegeneration, and therapeutic strategies targeting this pathway..
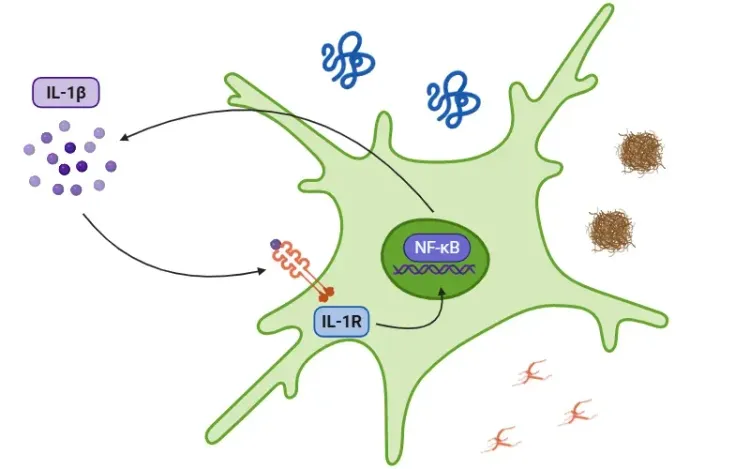
Interleukin-1 Beta (IL-1β) and Neurodegenerative Diseases
The role of IL-1beta in neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS).