TNF-α (TNF-alpha) & Microglia in Neurodegenerative Diseases
An overview of the function of tumor necrosis factor-alpha (TNF-α) in microglia and its contribution to the progression of neurodegeneration.
What is the role of TNF-α in the central nervous system (CNS)?
Tumor necrosis factor-alpha (TNF-α) is a pro-inflammatory cytokine that plays a crucial role in regulating inflammation and immune responses. In the central nervous system (CNS), TNF-α has a dual function, contributing positively under healthy conditions, but potentially causing harm when dysregulated in diseased states.
Under homeostatic conditions, TNF-α supports:
- Neuroplasticity
- Myelination
- Tissue repair
However, elevated TNF-α levels can lead to:
- Excitotoxity
- Chronic inflammation
- Breakdown of the blood-brain barrier (BBB)
TNF-α exists in both soluble and transmembrane forms, and acts through two main receptors:
- TNF receptor type-1 (TNFR1)
- TNF receptor type-2 (TNFR2)
Its expression is tightly regulated by various transcription factors, notably nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). When activated, NF-κB:
- Promotes TNF-α production
- Triggers the expression of other pro-inflammatory cytokines, such as IL-6 and IL-1β
- Creates a positive feedback loop that sustains and amplifies the inflammatory response (Gonzalez Caldito, 2023).
See: What is NF-κB (Nuclear Factor Kappa B)?
The main source of TNF-α during neuroinflammation is microglia, the resident immune cells of the CNS. Astrocytes can also contribute to its release under certain conditions (Olmos, 2014).
See: TNF-α & Astrocytes in Neurodegenerative Diseases
In the context of neurodegenerative diseases, microglial reactivity, along with other states like microglial senescence and the senescence-associated secretory phenotype (SASP), drives the chronic overproduction of TNF-α. Additional inflammatory cytokines, such as interferon-gamma (IFN-ϒ), further stimulate microglia, reinforcing the inflammatory cycle. This sustained neuroinflammatory state is implicated in the progression of diseases such as:
- Alzheimer's disease (AD)
- Parkinson’s disease (PD)
- Amyotrophic lateral sclerosis (ALS)
Therapeutic strategies targeting TNF-α have shown potential in treating CNS autoimmune diseases, and may offer a way to modify the neuropathological features of neurodegenerative diseases (Torres-Acosta, 2020; Gonzalez Caldito, 2023). However, such treatments are not without complications. In some cases, anti-TNF-α therapies have exacerbated conditions like multiple sclerosis (MS) (Fresegna, 2020). This paradox likely stems from the dual role of TNF-α: it can be both neurotoxic and neuroprotective depending on context and receptor involvement.
Ongoing research into the temporal dynamics of TNF-α expression, especially in early versus late stages of disease progression, may offer crucial insights (Guidotti, 2021). Understanding when and how to modulate TNF-α could determine whether such interventions are helpful or harmful.
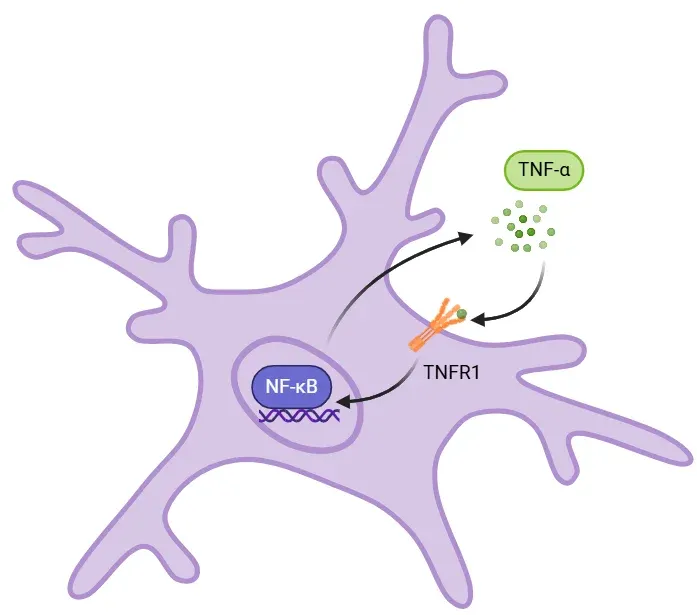
TNF-α-NF-κB Feedback Loop in Inflammation.
Binding of soluble TNF-α to TNFR1 activates NF-κB, which enters the nucleus and promotes transcription of proinflammatory cytokines like IL-6, IL-1β, and TNF-α. This creates a positive feedback loop that amplifies inflammation. Sustained TNF-α signaling can cause tissue damage and excitotoxity, contributing to chronic inflammation in neurodegenerative diseases.
How is TNF-α implicated in neurodegenerative diseases such as AD, PD, and ALS?
Alzheimer's Disease (AD)
AD is a neurodegenerative disease marked by progressive cognitive decline, memory impairment, and the accumulation of amyloid-beta (Aβ) plaques and tau neurofibrillary tangles. Increasing evidence suggests that inflammatory cytokines, including TNF-α, play a pivotal role in AD pathology.
In patients with AD, elevated levels of cytokines - notably TNF-α, IL-1β, and IL-6 - have been detected in:
- The blood
- Cerebrospinal fluid (CSF)
However, findings are not always consistent, likely due to differences in disease stage (Brosseron, 2014).
Despite these inconsistencies, several trends have emerged:
- Cytokine levels (particularly TNF-α) tend to increase as AD progresses
- Cytokine-to-receptor ratios may serve as better biomarkers than cytokine levels alone
- Elevated cytokines may reflect a higher risk of developing AD
A major contributor to this inflammatory environment is microglial dysfunction. In AD models with Aβ accumulation:
- Aged microglia display reduced phagocytic ability
- There is increased TNF-α and IL-1β expression
- Microglia reactivity promotes a feedback loop:
- Aβ plaques activate microglia
- Microglia release TNF-α
- TNF-α intensifies inflammation and neuronal damage
- Damage leads to further Aβ accumulation
This cycle perpetuates the neurodegenerative process, linking inflammation directly to disease progression (Hickman, 2008).
Therapeutic efforts to inhibit TNF-α in AD have shown mixed results:
- Etanercept (TNF-α inhibitor) via perispinal administration showed cognitive improvements in small open-label trials (Tobinick, 2008)
- Peripheral administration of etanercept showed no significant benefit (Butchart, 2015)
- A Phase 1b trial for XPro1595, targeting soluble TNF-α, has shown promise (NCT03943264)
This shift toward selectively inhibiting soluble TNF-α offers a more targeted strategy with potentially fewer side effects.
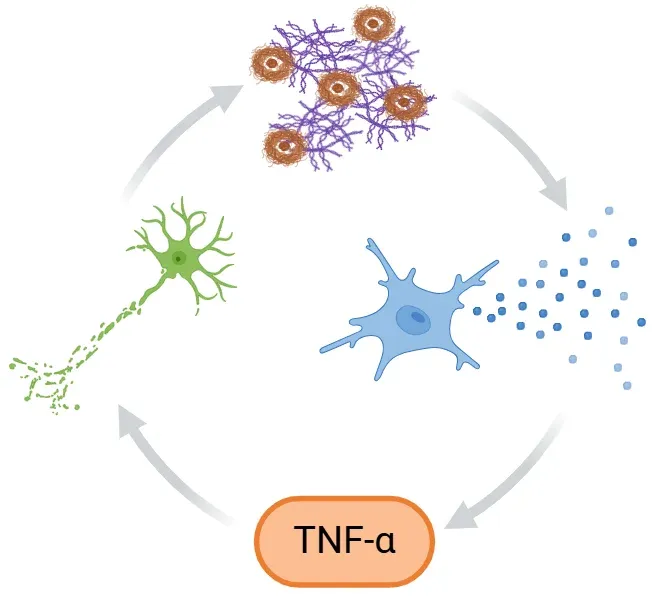
Inflammatory Feedback Loop in Alzheimer's Disease.
In Alzheimer's disease, Aβ plaques and tau tangles activate microglia, triggering release of pro-inflammatory cytokines like TNF-α. This amplifies neuroinflammation and neurodegeneration. At the same time, impaired microglial clearance leads to further accumulation of toxic proteins, reinforcing the cycle and accelerating disease progression.
Parkinson’s Disease (PD)
PD is a progressive neurodegenerative disease characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNc), resulting in hallmark motor symptoms such as tremor, rigidity, and bradykinesia. It also includes non-motor symptoms like cognitive impairment and sleep disturbances.
Chronic neuroinflammation is a key component of PD pathology. Evidence includes:
- Microglial reactivity and elevated TNF-α in postmortem SN samples and serum (Collins, 2012)
- Increased TNF-α in serum and CSF in PD animal models
- Presence of TNFR1 on dopaminergic neurons in the SNc
In PD patients, increased activation of NF-κB, a transcription factor for pro-inflammatory genes, has also been observed (Mogi, 2007). This activation reinforces cytokine expression and microglial reactivity.
Therapeutic studies in animal models support the benefit of TNF-α inhibition:
- Thalidomide, a TNF-α synthesis inhibitor, protected against MPTP-induced dopamine depletion (Ferger, 2004)
- TNF-α-deficient mice showed reduced striatal dopamine loss
- XENP345, a TNF-α blocker, reduced dopaminergic neuron death (McCoy, 2006)
These findings highlight TNF-α as a potential therapeutic target to modulate inflammation and slow disease progression in PD.
Amyotrophic Lateral Sclerosis (ALS)
ALS is a progressive neurodegenerative disease that primarily targets motor neurons, leading to muscle weakness, difficulty speaking and swallowing, spasticity, paralysis, and eventual respiratory failure.
Neuroinflammation contributes significantly to disease progression, with elevated TNF-α levels observed in:
- Plasma and serum of ALS patients
- Animal models, particularly the superoxide dismutase 1 (SOD1) transgenic mouse (Olmos, 2014; Vu, 2017)
In these models:
- NF-κB activation in spinal microglia is consistently observed (Frakes, 2014)
However, the role of TNF-α in ALS is more complex. Gene knockout studies in SOD1 model (removing TNF-α) did not significantly alter disease course, suggesting that while TNF-α may contribute to inflammation, it is not the primary driver of motor neuron loss in some contexts (Gowing, 2006).
Despite this, TNF-α inhibition has shown benefits. Thalidomide and lenalidomide improved motor function and reduced motor neuron loss in SOD1 mice (Kiaei, 2006) However, a Phase 2 trial in humans found no clinical benefit and reported notable side effects (Stommel, 2009).
These mixed results underscore the importance of:
- Timing: Early intervention may be more effective
- Stage-specific targeting: TNF-α may play different roles at different points in disease progression (Guidotti, 2021)
In conclusion, TNF-α plays a central role in neuroinflammation and neurodegeneration across diseases like AD, PD, and ALS. Despite differences in their underlying mechanisms, inflammatory processes driven by TNF-α appear to be a common factor in disease progression. While therapies tarting TNF-α show promise in preclinical models, clinical results have been inconsistent, highlighting the need for a deeper understanding of TNF-α’s role. Future research should focus on refining targeted treatments that modulate TNF-α signaling, potentially offering therapies to slow or halt the progression of these neurodegenerative diseases.
What evidence suggests that modulating TNF-α may slow or prevent neurodegenerative diseases?
Given its pivotal role in driving neuroinflammation, TNF-α is central to the progression of many neurodegenerative diseases, including AD, PD, and ALS. Chronic elevation of TNF-α in the CNS leads to neuronal damage and toxic protein accumulation, which together contribute to disease progression. As such, modulating TNF-α has emerged as a promising strategy to slow or even halt the progression of these diseases.
Anti-TNF-α therapies, successful in treating a range of autoimmune and inflammatory diseases, have drawn attention for their potential in neurodegenerative diseases (Gonzalez Caldito, 2023). Drugs such as etanercept (Enbrel®) and infliximab (Remicade®), widely used in treating conditions like rheumatoid arthritis and other peripheral inflammatory diseases, have limited effectiveness in CNS disorders due to poor BBB penetration (Decourt, 2017). Other TNF-α inhibitors face similar challenges in accessing the CNS:
- Adalimumab (Humira®)
- Golimumab (Simponi®)
- Certolizumab pegol (Cimzia®)
This challenge in TNF-α inhibition in CNS diseases is compounded by the risk of serious side effects, including:
- Increased susceptibility to infections
- Risk of demyelinating diseases
- Potential worsening of diseases like MS
For example, in MS, treatments like lenercept have worsened disease outcomes (Maguire, 2021). The dual roles of TNF-α in neuroprotection and inflammation underscore the need for more selective targeting. Research on TNFR1 and TNFR2 has highlighted that selectively targeting TNFR1, while sparing TNFR2, may offer a safer and more effective approach for treating MS (Fresegna, 2020).
In response to these limitations, development of selective TNF-α inhibitors that specifically target TNFR1 for CNS use, such as atrosab and atrosimab, have demonstrated promising results in preclinical models of neuroinflammation, such as experimental autoimmune encephalomyelitis (EAE) (Richter, 2021). Similarly, a Phase 1b trial for XPro1595, a TNF-α inhibitor selective for soluble TNF-α has shown promising results in AD (NCT03943264). These therapies aim to offer a more targeted approach, improving safety and efficacy for neurodegenerative diseases.
While preclinical findings are promising, further clinical trials are necessary to establish the safety and effectiveness of these selective TNF-α blockers in humans. These studies will be crucial for determining whether these therapies can provide benefits for patients with neurodegenerative diseases. As research continues, the development of TNF-α therapies tailored for the CNS may offer an important approach to slowing or preventing neurodegeneration, improving outcomes for patients and advancing the field of neuroinflammation treatment (Decourt, 2017; Hampel, 2020; Zahedipour, 2022).
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on TNF-α and microglia in neurodegenerative diseases and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
TNF-α & (TNF-alpha) Astrocytes in Neurodegenerative Diseases
An overview of TNF-α signaling in astrocytes, its role in neurodegeneration, and therapeutic strategies targeting this pathway..
Microglia, Astrocytes & Tau in Neurodegenerative Diseases
How glial-driven neuroinflammation fuels tau aggregation, propagation, and neuronal loss in Alzheimer’s disease and other tauopathies.
Microglia, Astrocytes & α-Synuclein in Parkinson’s Disease
How α-synuclein influences microglia and astrocytes in Parkinson’s disease and other synucleinopathies.
Mitochondrial Dysfunction in Microglia & Astrocytes
The role of mitochondrial dysfunction in microglia and astrocytes in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and ALS.
Microglial Senescence and Neurodegenerative Diseases
An overview of microglial senescence and its role in neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD).
Microglia Morphology in ALS, Alzheimer's Disease & Parkinson's Disease
An overview of microglial morphological analysis and the applications to neurodegenerative disease research and drug discovery & development.
Amyloid-β & Inflammatory Microenvironment in Alzheimer's Mice
We have analyzed the complex spatial relationships between β-amyloid plaques, activated & resting microglia, and astrocytes in an APP/PS1 transgenic model.