Microglia, Astrocytes & α-Synuclein in Parkinson’s Disease
How α-synuclein influences microglia and astrocytes in Parkinson’s disease and other synucleinopathies.
This resource describes:
How do α-synuclein, microglia, and astrocytes contribute to the development of PD?
Clinical and Pathological Features of PD
Parkinson’s disease (PD) is the most common movement-related neurodegenerative disease and the second most prevalent neurodegenerative disease overall, affecting 2-3% of individuals over 65 years-old (Poewe, 2017). As global life expectancy rises, the burden of PD is expected to increase significantly.
The disease is classified among the α-synucleinopathies (or synucleinopathies), a heterogeneous group of diseases characterized by the accumulation of misfolded α-synuclein. α-Synuclein consists of three domains: an N-terminus, a non-amyloid-β component (NAC) domain, and a C-terminus. Under normal conditions, α-synuclein is involved in vesicle trafficking and neurotransmitter release, particularly through its role in SNARE complex assembly (Calabresi, 2023). However, when its synthesis or clearance is disrupted, α- synuclein can misfold and aggregate into oligomers and fibrils. These aggregated forms of α-synuclein propagate in a stereotypical pattern across brain regions (Braak, 2003), forming Lewy bodies (LBs) and Lewy neurites (LNs). Interestingly, the presence and distribution of Lewy pathology do not always correlate with clinical severity, suggesting that α-synuclein aggregation is one component of a broader pathological process. While the trigger for α-synuclein aggregation remains uncertain, factors such as neuroinflammation, mitochondrial dysfunction, and oxidative stress are believed to contribute to the process (Borsche, 2021; Chen, 2023).
PD presents with both motor and non-motor symptoms. The hallmark motor features–bradykinesia, resting tremor, and rigidity–are primarily attributed to progressive dopaminergic neuron loss in the substantia nigra pars compacta (SNc). Non-motor symptoms include depression, cognitive impairment, sleep disturbances, and gastrointestinal dysfunction. Early diagnosis of PD is challenging because motor symptoms typically appear only after significant dopaminergic neuron loss has occurred, and diagnosis is further complicated by overlapping clinical features with other disorders, such as multiple system atrophy (MSA) and progressive supranuclear palsy (PSP). These diagnostic challenges underscore the need for validated early-stage biomarkers to improve disease detection and therapeutic stratification.
The underlying mechanisms of neurodegeneration in PD remain poorly understood, though a variety of factors, including genetic, environmental, and aging factors have been linked to disease pathogenesis. Additionally, dysregulation of both innate and adaptive immune responses may play a critical role in the pathogenesis of PD, and, therefore, targeting inflammatory pathways for PD treatment has gained significant attention (Li, 2021; Chen, 2023).
Role of Neuroinflammation in PD
Neuroinflammation is increasingly recognized as a key contributor to PD pathogenesis. Chronic activation of immune responses within the central nervous system (CNS), particularly involving glial cells such as microglia and astrocytes, plays a central role in disease progression. A major function of astrocytes and microglia involves the immune response, although they also contribute to blood-brain barrier (BBB) function, CNS homeostasis, phagocytosis, synaptic function, ion balance, and metabolic support. In response to environmental insults, like toxins, infections, or injury, glial cells adopt a reactive, pro-inflammatory phenotype and can release pro-inflammatory cytokines, such as interleukin-1 β (IL-1β) and tumor necrosis factor alpha (TNF-α).
A major mediator of glial-driven inflammation is the Nod-like receptor protein 3 (NLRP3) inflammasome, a multiprotein complex that responds to cellular stress signals, including the accumulation of misfolded and aggregated proteins like α-synuclein. Once activated, the NLRP3 inflammasome promotes the release of pro-inflammatory cytokines, including IL-1β and interleukin-18 (IL-18), exacerbating neuroinflammation and neuronal damage.
See: What is an inflammasome? & What is NLRP3?
Evidence supports the early involvement of neuroinflammation and microglial reactivity in the SN of PD patients, suggesting a window for therapeutic intervention (Gerhard, 2006; Edison, 2013). Microglial reactivity in PD is associated with a reduction in triggering receptor expressed on myeloid cells 2 (TREM2), a receptor expressed in microglia that is implicated in neuroinflammation and phagocytosis (Huang, 2021). Studies show that increasing levels of TREM2 enhances autophagy, decreases dopaminergic neuron death, and improves motor ability in PD mouse models (Huang, 2021). Furthermore, TREM2 deficiency has been shown to activate the NLRP3 inflammasome, whose activation has been detected in the SNc of PD brains (Gordon, 2018; Huang, 2024). In PD mouse models, NLRP3 inhibition reduces dopaminergic neuron loss, diminishes the secretion of pro-inflammatory cytokines, improves motor function, and prevents α-synuclein pathology by relieving autophagy (Gordon, 2018; Lee, 2019; Ou, 2021).
For an in-depth review of the role of TREM2 on microglial function, see: TREM2 and Microglia
Aggregated α-synuclein plays a central role in this inflammatory cascade (Wang, 2016). α-Synuclein activates microglia via toll-like receptors (TLRs), particularly TLR2 and TLR4, leading to NF-κB activation and the transcription of pro-inflammatory cytokines, such as IL-1β and TNF-α (Karpenko, 2018; Feng, 2019). NF-κB also regulates the expression of the NLRP3 inflammasome in microglia, resulting in further cytokine release (Wang, 2024). These cytokines not only damage neurons directly, but also stimulate astrocytes to become reactive and adopt a pro-inflammatory state. Moreover, α-synuclein interacts with mitochondrial components, impairing mitochondrial function, dynamics and biogenesis, inducing reactive oxygen species (ROS), and contributing to cell death (Grünewald, 2019; Jeon, 2020). This feed-forward loop amplifies neuroinflammation in PD and contributes to the progressive loss of dopaminergic neurons in the SNc (Calabresi, 2023).
For more on the role of mitochondrial dysfunction in neurodegenerative diseases, see: Mitochondrial Dysfunction in Microglia & Astrocytes
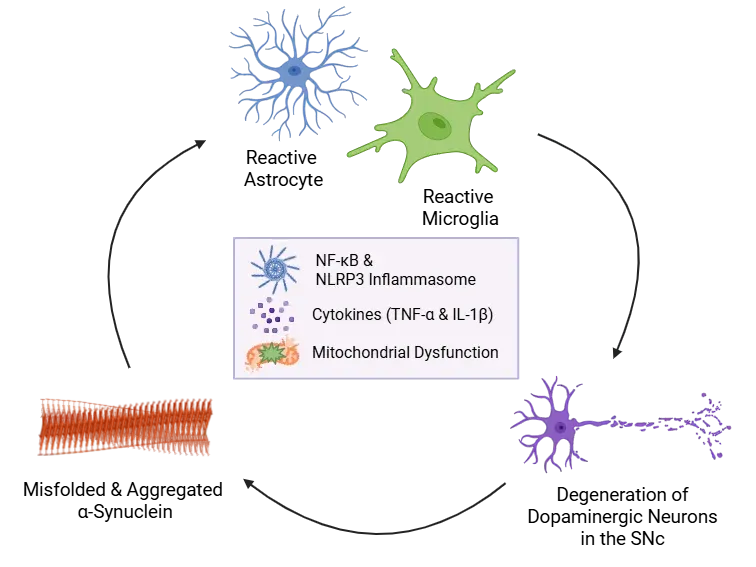
α-Synuclein Aggregation Drives Neuroinflammation.
Misfolded α-synuclein activates astrocytes and microglia, triggering NF-κB signaling, cytokine release (e.g. IL-1β and TNF-α), and NLRP3 inflammasome priming. Inflammasome activation amplifies inflammation, promotes further α-synuclein aggregation, mitochondrial dysfunction, and neuronal damage - creating a self-perpetuating cycle of neuroinflammation and dopaminergic loss in the SNc.
In addition to the innate immune system, the adaptive immune response is also implicated in PD. Cytotoxic T cells have been observed in the SN of PD patients, suggesting peripheral immune infiltration into the CNS. Both CD4+ and CD8+ T cells have been detected in affected brain regions in patient samples and animal models (Brochard, 2009). Interestingly, NLRP3 inhibition reduces CD4+ and CD8+ T cell infiltration into the SNc, supporting the role of the NLRP3 inflammasome in adaptive inflammation (Grotemeyer, 2023). Notably, depletion of CD4+ T cells in mouse models reduces dopaminergic neuron death, further supporting the pathogenic role of adaptive immunity in PD (Brochard, 2009).
Research also highlights the potential contributor of the gut-brain axis in modulating neuroinflammation (Houser, 2017). Alterations in gut microbiota composition and intestinal permeability may promote inflammation via circulating inflammatory mediators from the intestine. These findings suggest that systemic inflammation originating outside the CNS may play a role in PD initiation or progression, and may reflect one of the earliest manifestations of PD pathology (Houser, 2017).
Furthermore, aging–the primary risk factor for PD–is associated with microglial senescence and chronic neuroinflammation. Senescent microglia accumulate in the aging brain, including the SNc, and adopt a reactive, pro-inflammatory phenotype (Shaerzadeh, 2020). Senescent microglia exhibit an increased release of ROS and pro-inflammatory cytokines (e.g. TNF-α and IL-1β), the accumulation of iron, and reduced phagocytic ability (Hong, 2024). Importantly, older mice exhibit higher levels of α-synuclein and slower α-synuclein clearance, likely due to age-related dysfunction in the autophagy-lysosome system (Hong, 2024). This age-related reduced α-synuclein clearance capacity may critically affect PD pathology, sustaining neuroinflammation and dopaminergic neuron vulnerability.
In conclusion, neuroinflammation represents a central mechanism in PD pathology with both innate and adaptive immune components contributing to disease progression. Early detection through neuroinflammatory biomarkers may enable more accurate diagnosis (Chen, 2023; Youssef, 2023). These biomarkers could also guide the development of targeted, immunomodulatory therapies aimed at halting or slowing disease progression.
Genetic Factors Influencing α-Synuclein and Glial Interaction
Genetic contributions to PD are well recognized, including several known mutations in SNCA, DJ-1, PRKN, PINK1, GBA1, and LRRK2 genes. SNCA, the gene encoding α-synuclein, is a major contributor to familial PD. SNCA mutations (e.g. A53T and A30P), as well as duplications or triplications, are associated with autosomal dominant forms of the disease. Further, variants in SNCA are among the most significant genetic risk factors identified in genome-wide association studies of idiopathic PD (Edwards, 2010). Genetic contributors to PD are also linked to mitochondrial dysfunction and neuroinflammation (Saijo, 2009; Yao, 2023). For example, mutations of LRRK2 (leucine-rich repeat kinase 2) are associated with mitochondrial dysfunction, including dysfunction in lysosomal pathways and impaired mitophagy (Borsche, 2021). In rodent models, LRRK2 knockout reduces microglial activation and neuronal loss, while pharmacological LRRK2 inhibition lowers both neuroinflammation and neurodegeneration (Daher, 2014, 2015). Consequently, the identification of a “mitochondrial subtype” of PD through biomarker profiling has prompted the development of gene-specific therapies, potentially allowing for more personalized treatment strategies (Borsche, 2021).
How are rodent models used to study the interaction between α-synuclein pathology and neuroinflammation?
Several rodent models of PD have been developed, including transgenic, knock-in, knock-out, neurotoxin, α-synuclein preformed fibrils (PFFs)-induced, and viral vector-induced approaches. Several of these models recapitulate some of the key pathological features and behavioral deficits associated with PD. However, animal models that go beyond the primary pathological hallmarks and elucidate the role of neuroinflammation and mitochondrial dysfunction in PD are a particularly valuable resource (Dovonou, 2023).
- Neurotoxic models are widely used in PD research, particularly MPTP, which induces acute dopaminergic neurotoxicity and significant motor impairment. One advantage of this model is that it produces robust microglial activation, NLRP3 inflammasome activation, and pro-inflammatory cytokine release in the SN (Lee, 2019). Similarly, 6-hydroxydopamine (6-OHDA) lesions of the dopamine system induced via intracranial injection result in both motor impairments and non-motor phenotypes. However, the relevance of neurotoxic models is limited by the absence of α-synuclein aggregation or the formation of Lewy body-like inclusions. Despite these limitations, neurotoxic models are suitable for studying oxidative stress processes and neuroinflammation (Dovonou, 2023).
- Transgenic mice overexpressing human wild-type (WT) or PD-linked mutations in different genes under various promotors are also widely utilized. These models are important for investigating the function of PD-linked genes. For example, transgenic models expressing WT, A53T, A30P or E46K SNCA mutations have been used to recapitulate PD pathology in vivo. Notably, some transgenic models, such as the G2019S LRRK2 overexpression mouse model demonstrates neuroinflammation and mitochondrial dysfunction (Dovonou, 2023). However, despite their advantages, it has been suggested that models that directly induce α-synuclein misfolding and seeding may more closely replicate PD-relevant pathological mechanisms (Calabresi, 2023).
- Injecting α-synuclein PFFs into specific brain regions is a pathologically relevant approach. These fibrils seed endogenous α-synuclein and propagate pathology, mimicking the spatiotemporal progression of PD pathology. Notably, PFF injections trigger strong microglial responses and pro-inflammatory signalling, directly linking α-synuclein misfolding to immune activation, while also reducing TREM2, increasing TLR4, and enhancing cytokine release, supporting the relationship between α-synuclein pathology and neuroinflammation (Thomsen, 2021; Niskanen, 2025).
- Viral vector-based models, particularly those utilizing adeno-associated viruses (AAVs), enable localized human WT or mutated α-synuclein expression in specific brain regions, such as the SNc. Using this approach, it is possible to trigger extensive PD-like pathology involving α-synuclein aggregation, neuronal dysfunction, and cell death that develops progressively over time (Ulusoy, 2010; Lundblad, 2012; Bourdenx, 2015; Phan, 2017). In addition, AAV-induced overexpression of human α-synuclein leads to microglial activation in the SN, recruitment of the adaptive immune system, and the production of pro-inflammatory cytokines (Theodore, 2008; Sanchez-Guajardo, 2010). Thus, AAV models provide valuable insights into PD pathology, including neuroinflammation, while enabling region-specific targeting.
In summary, rodent models remain essential for studying the interplay between α-synuclein pathology and neuroinflammation in PD. While each model has both advantages and limitations, they collectively provide valuable insight into disease mechanisms and therapeutic targets. Notably, α-synuclein PFF and viral vector models closely replicate key pathological and immune features, supporting their use in preclinical testing of disease-modifying therapies.
What therapeutic strategies are being developed to target α-synuclein and neuroinflammation?
Current treatment for PD primarily manages motor symptoms using dopaminergic therapies, with levodopa (L-DOPA) remaining the gold standard. Regimens often involve combining levodopa with dopamine agonists, COMT inhibitors, or MAO-B inhibitors to optimize dopaminergic availability. However, while effective initially, long-term use is associated with complications such as dyskinesias, motor fluctuations, and a range of non-motor side effects.
In contrast to these symptomatic treatments, disease-modifying strategies are a major research focus. Among them, immunotherapies and anti-aggregation agents targeting α-synuclein are particularly prominent (Chatterjee, 2019; Alfaidi, 2024). These approaches aim to interfere with the accumulation and pathological spread of α-synuclein, as well as the inflammatory responses elicited in glial cells.
Immune-Based Approaches
Both passive and active immunization strategies have shown early promise. Active immunisation agents are designed to provoke an immune response against α-synuclein. Peptide-based vaccines, AFFITOPE® PD01A and PD03A, are active vaccines designed to generate an immune response against the oligomeric forms of α-synuclein, aiming to reduce its aggregation and spread. These vaccines have completed a series of Phase 1 clinical trials demonstrating safety and target engagement, with PD03A generating lower antibody titres than PD01A (Volc, 2020; Poewe, 2021). PD01A has since been reformulated and renamed ACI-7104.056 and is currently being investigated in a Phase 2 trial (NCT06015841). UB-312, another synthetic peptide-based vaccine designed to induce active immunity against pathogenic α-synuclein, is currently in a Phase 1b trial (NCT05634876) following a Phase 1a trial that showed a significant reduction of α-synuclein in CSF in a subset of patients (Eijsvogel, 2024).
In the realm of passive immunisation, PRX002 (prasinezumab), a humanized IgG1 monoclonal antibody targeting the C-terminal of aggregated α-synuclein, has demonstrated high affinity for pathological forms over monomers. Phase 1b data showed it was safe, well tolerated, and reduced free α-synuclein in serum (Jankovic, 2018). While a Phase 2 trial (NCT03100149) did not meet its primary efficacy endpoint, exploratory analyses suggested motor-symptom slowing in specific patient subgroups (Pagano, 2022; 2024). Other monoclonal antibodies have had mixed results. BIIB054 (cinpanemab), a monoclonal antibody that targets aggregated α-synuclein, failed to show clinical or imaging-based efficacy in a Phase 2 trial (Lang, 2022). In contrast, BAN0805 (exidavnemab), a monoclonal antibody designed to selectively bind and eliminate aggregated α-synuclein, showed dose-dependent reductions in plasma α-synuclein and good tolerability in Phase 1. It is currently in Phase 2a evaluation (NCT06671938) (Boström, 2024).
Targeting the NLRP3 neuroinflammatory pathway is another promising strategy. VTX3232, a brain-penetrant NLRP3 inflammasome inhibitor, aims to suppress microglial activation and downstream cytokine release. It is currently in a Phase 2a trial in early-stage PD (NCT06556173). Similarly, VENT-02, a brain-penetrant NLRP3 inhibitor, is currently in a Phase 1b trial, following a phase 1a trial, which showed significant inhibition of IL-1β in the blood (NCT06822517).
Obstacles and Advances in Developing Disease-Modifying Therapies
One of the central challenges in developing disease-modifying therapies is the difficulty in diagnosing PD early and accurately. In its prodromal stages, PD symptoms often overlap with those of other Parkinsonian disorders, complicating trial stratification and therapeutic timing. The absence of a reliable biomarker to track disease progression further limits the ability to assess treatment efficacy.
To address these challenges, techniques such as advanced neuroimaging and fluid biomarkers, such as neurofilament light chain, are being explored to support early diagnosis, track neurodegeneration, and improve trial design. Significant efforts have also been made to identify biomarkers linked to α-synuclein and neuroinflammation. These biomarkers may offer a therapeutic window before extensive neuronal loss occurs. Biomarkers based on neuroinflammatory markers appear promising for early detection of PD, and combining multiple markers, including inflammatory ones, may enhance diagnostic accuracy (Chen, 2023; Youssef, 2023).
Future PD therapies will likely combine α-synuclein -targeted treatments with agents that modulate inflammation and mitochondrial function. Personalized approaches based on genetic or biomarker profiles may improve efficacy, while advances in drug delivery and early intervention could enhance outcomes.
Our team would be happy to answer any questions about microglia, astrocytes & α-synuclein in Parkinson’s disease or provide specific information about the PD models we use for therapeutic efficacy studies.
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on Neuroinflammation and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
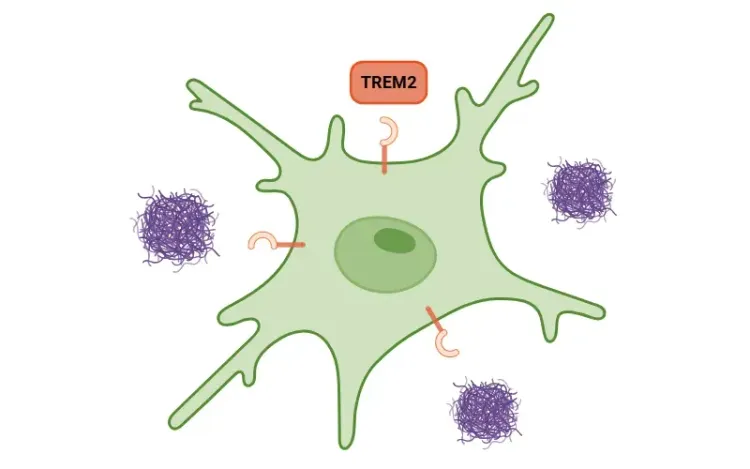
TREM2, Microglia and Neuroinflammation
An overview of TREM2, its role in microglia, links to neurodegenerative diseases, and potential treatment implications.
Microglia, Astrocytes & Tau in Neurodegenerative Diseases
How glial-driven neuroinflammation fuels tau aggregation, propagation, and neuronal loss in Alzheimer’s disease and other tauopathies.
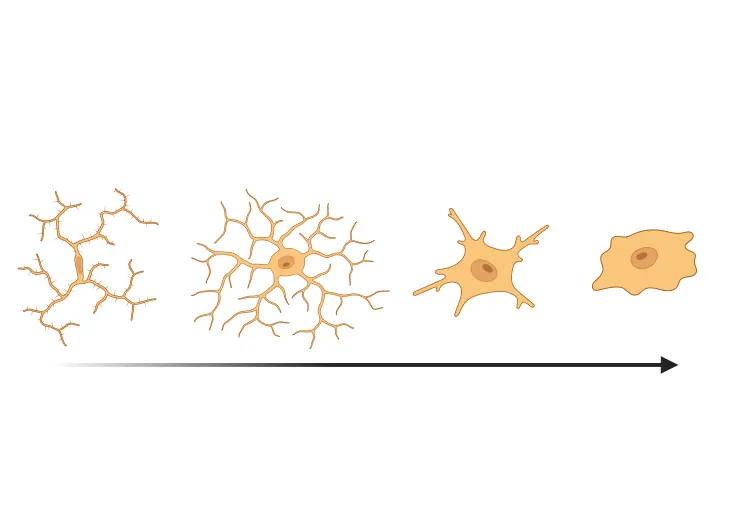
Microglial Activation in an α-Synuclein PFF Mouse Model
We have quantified microglial activation, based on morphology, in an α-synuclein preformed fibril (PFF) seeding & spreading mouse model of Parkinson’s disease.
AAV α-Synuclein Models for Parkinson's Disease Drug Development
Overview of adeno-associated virus (AAV) induced α-synuclein expression in mouse & rat models for use in preclinical studies of disease-modifying therapeutics.
Autophagy, Parkinson's Disease, and Dopaminergic Neurons
An overview of how impaired autophagy can lead to pathologic changes and neurodegeneration in dopaminergic neurons in Parkinson’s disease.
Mitochondrial Dysfunction in Microglia & Astrocytes
The role of mitochondrial dysfunction in microglia and astrocytes in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and ALS.
Microglial Senescence and Neurodegenerative Diseases
An overview of microglial senescence and its role in neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD).
NLRP3 Inflammasome and Neurodegenerative Diseases
An overview of the NLRP3 inflammasome and its role in neurodegenerative diseases, including Alzheimer's disease, Parkinson’s disease, and ALS.
TNF-α (TNF-alpha) & Microglia in Neurodegenerative Diseases
An overview of the function of tumor necrosis factor-alpha (TNF-α) in microglia and its contribution to the progression of neurodegeneration.