Mitochondrial Dysfunction in Microglia & Astrocytes
The role of mitochondrial dysfunction in microglia and astrocytes in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and ALS.
What is the role of mitochondria in microglia and astrocytes?
Mitochondrial Function and Dynamics
Mitochondria are essential organelles that support a wide range of cellular functions. Known as the “powerhouses” of the cell, mitochondria generate adenosine 5’ triphosphate (ATP), the primary energy source for cellular processes, mainly through oxidative phosphorylation (OXPHOS) via the electron transport chain (ETC), which comprises a series of protein complexes (Tönnies, 2017). However, their roles extend well beyond energy production. Mitochondria are also involved in lipid and amino acid metabolism, calcium (Ca2+) homeostasis, and programmed cell death (PCD), often referred to as apoptosis (Bose, 2016). Moreover, they modulate oxidative stress by balancing the production of reactive oxygen species (ROS) with antioxidant defenses.
Mitochondria are highly dynamic and undergo continuous morphological remodeling through processes of fusion and fission. These dynamic changes are critical for maintaining mitochondrial function and preventing apoptosis. Key proteins involved in mitochondrial dynamics include dynamin-related protein 1 (Drp1) for fission, and mitofusins (Mfn1/2) and optic atrophy 1 (OPA1) for fusion. Disruption in the balance of these processes can result in mitochondrial fragmentation, reduced ATP production, and increased cellular stress. In addition, defective fission and fusion can impair mitochondrial transport, contributing to synaptic dysfunction and neurodegeneration.
Mitochondrial Dysfunction and Glial-Mediated Neuroinflammation
Mitochondrial dysfunction may result from various factors, including aging and mutations in mitochondrial DNA (mtDNA), leading to impaired ATP production, increased ROS levels, structural abnormalities, oxidative stress, and apoptosis (Tönnies, 2017; Wang, 2020). While physiological levels of ROS serve as signaling molecules, excessive ROS disrupt mitochondrial integrity and function (Kumar, 2012). Damaged mitochondria that are not adequately cleared through mitophagy may release mitochondrial-derived damage-associated molecular patterns (DAMPs), which trigger inflammatory signaling pathways through the activation of innate immune receptors (Salmina, 2021; Lin, 2022).
Microglia and astrocytes play critical roles in the central nervous system (CNS) immune response and are key responders to mitochondrial DAMPs. Microglia recognize these danger signals through pattern recognition receptors, activating transcription factors, such as NF-κB, which drive the release of pro-inflammatory cytokines like interleukin-1β (IL-1β) and tumor necrosis factor alpha (TNF-α). These cytokines exacerbate oxidative stress, disrupt the blood-brain barrier (BBB), and promote neuronal injury (Wang, 2020). In both microglia and astrocytes, mitochondrial dysfunction impairs neuroprotective functions and sustains chronic inflammation. As such, a vicious cycle is created, whereby mitochondrial damage activates glial cells, and the resulting inflammatory mediators further damage mitochondria, amplifying neurodegeneration (Jeon, 2020; Yan, 2020).
Importantly, mitochondrial dysfunction may precede neuroinflammation in neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS), positioning mitochondrial dysfunction as a possible driver of disease pathology (Swerdlow, 2014). Investigating mitochondrial changes in glial cells, in addition to neurons, may provide a broader understanding of early disease mechanisms and facilitate the development of novel therapeutic strategies (Lin, 2022).
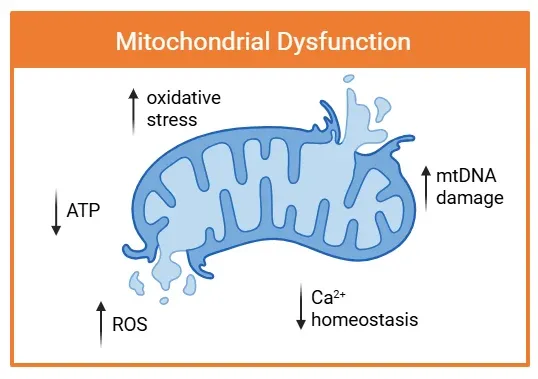
Mitochondrial dysfunction is characterized by structural damage and impaired function. Arrows indicate key pathological changes, including increased ROS production, oxidative stress, and mtDNA damage. These impairments coincide with decreased ATP generation and disrupted Ca2+ homeostasis, contributing to neuroinflammation and neurodegenerative disease progression.
What is the link between mitochondrial dysfunction in microglia & astrocytes and AD, PD, and ALS?
Mitochondrial dysfunction in microglia and astrocytes is an emerging area of interest in the study of neurodegenerative diseases. Impaired mitochondrial function in these glial cells not only impairs their supportive roles, but also initiates and sustains neuroinflammation, creating a feedback loop that exacerbates neuronal injury.
Alzheimer’s Disease (AD)
AD is clinically characterized by progressive memory loss, cognitive decline, and behavioral changes. Its pathological hallmarks include the accumulation of amyloid-beta (Aβ) plaques, hyperphosphorylated tau neurofibrillary tangles, and neuronal loss. Notably, mitochondrial dysfunction, oxidative stress, and inflammation are among the earliest features contributing to AD progression (Swerdlow, 2014).
In AD, Aβ accumulation within mitochondria inhibits ETC complex-II and IV, reducing ATP production and increasing ROS (Kumar, 2015; Swerdlow, 2016). Aβ also disrupts mitochondrial protein import, damages mtDNA, and impairs CA2+ homeostasis, promoting apoptosis (Kumar, 2015). Furthermore, Aβ disrupts mitochondrial morphology and dynamics, including the regulation of fission and fusion (Wang, 2008; 2009). Dysregulation of Drp1 and OPA1, which mediate these dynamics, is linked to Aβ-induced neuronal toxicity (Wang, 2008; 2009). The impaired clearance of damaged mitochondria amplifies neurodegeneration (Kumar, 2015).
Preclinical models suggest mitochondrial-targeted interventions may offer therapeutic benefit. For example, OPA1 overexpression restores mitochondrial structure and function in AD models (Wang, 2008; 2020). Moreover, P110, a selective Drp1/Fis1 inhibitor, reduces mitochondrial fragmentation and both microglial & astrocytic activation, reducing the release of damaged mitochondria from microglia, and protecting neurons from cytokine-mediated injury (Joshi, 2019). These findings support the potential of targeting mitochondrial fission in microglia to attenuate neuroinflammation and neuronal damage.
Parkinson’s Disease (PD)
PD presents with motor symptoms including bradykinesia, rigidity, gait difficulties, and resting tremor, along with non-motor symptoms such as cognitive impairment and mood & sleep disturbances. It is characterized by the accumulation of α-synuclein (α-syn) in Lewy bodies and degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc). While most PD cases are sporadic, some are associated with genetic mutations in genes like DJ-1 (also known as PARK7) (Almikhalfi, 2020).
Both familial and sporadic PD exhibit mitochondrial defects (Grünewald, 2019). DJ-1 knockout rats display progressive dopaminergic neurodegeneration in the SN, motor deficits, and disruptions in mitochondrial pathways, such as ETC complex I (Almikhlafi, 2020). Astrocytes help degrade α-syn through the autophagy-lysosome pathway; however, excessive α-syn overwhelms this process, leading to further mitochondrial dysfunction. α-Syn interacts with mitochondrial components, including cytochrome C oxidase 1 (COX1), impairing mitochondrial function (Jeon, 2020). Mutations in ATP13A2 also disrupt mitochondrial and lysosomal pathways, contributing to α-syn accumulation (Grünewald, 2019).
Amyotrophic Lateral Sclerosis (ALS)
ALS is a progressive neurodegenerative disease affecting motor neurons, leading to muscle weakness, spasticity, and respiratory failure. While most cases are sporadic, familiar ALS is linked to mutations in genes such as C9orf72, SOD1, FUS, and TARDBP. Misfolded proteins, like mutant superoxide dismutase 1 (SOD1) and TDP-43, are hallmark features of ALS.
Abnormal mitochondrial morphology has been documented in ALS patients, mice, and cell culture models (Magrané, 2009). Mitochondrial transport and positioning at synapses of the neuromuscular junction are crucial for motor neuron function. Postmortem studies show reduced ETC complex activity in the spinal cords of ALS patients (Obrador, 2020). Dysregulation of mitochondrial sirtuins, such as SIRT1 and SIRT3, impairs mitochondrial biogenesis and function (Obrador, 2020). SIRT1 activates PGC-1α, which enhances mitochondrial function and ROS defense via SIRT3 (Song, 2013). Activating SIRT1 with resveratrol improves symptoms and survivial in the SOD1G93A ALS mouse model, while SIRT3 overexpression decreases the production of ROS, and protects against mitochondrial fragmentation and death in SOD1G93A motor neurons in culture (Han, 2012; Song, 2013).
TDP-43 also interacts with mitochondrial proteins, such as the voltage-dependent anion channel 1 (VDAC1), disrupting mitochondrial dynamics and mitophagy (Obrador, 2020). In induced pluripotent stem cells (iPSC)-derived C9orf72 motor neurons, oxidative stress increases with age, but can be partially alleviated by reducing oxidative stress (Lopez-Gonzalez, 2016).
In summary, while AD, PD, and ALS differ in presentation and pathology, they share a common mechanism: mitochondrial dysfunction that drives chronic glial activation and neuroinflammation. This bidirectional relationship creates a feedback loop that accelerates disease progression. Targeting this cycle may offer new therapeutic avenues.
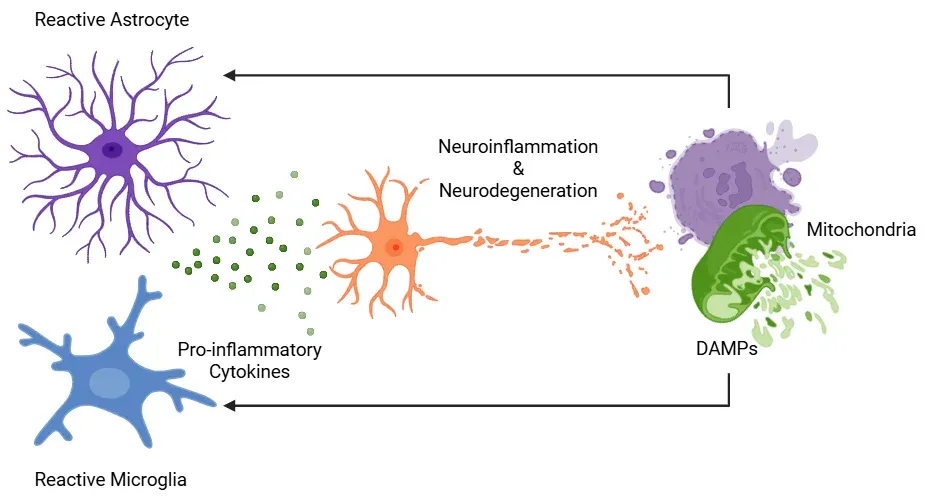
Neuroinflammation in neurodegenerative diseases is driven by the interaction between dysfunctional mitochondria and reactive glial cells. Damaged mitochondria release mitochondrial-derived DAMPs, which are recognized by microglia and astrocytes. In response, these glial cells adopt a reactive phenotype and secrete pro-inflammatory cytokines, further impairing mitochondrial function. This process establishes a self-perpetuating feedback loop wherein mitochondrial dysfunction and glial reactivity amplify each other, contributing to chronic neuroinflammation and progressive neuronal degeneration. Figure adapted from Picca et al. (Picca, 2022) under the Creative Commons Attribution License.
What are the therapeutic approaches targeting mitochondrial dysfunction in neurodegenerative diseases?
Given the central role of mitochondrial dysfunction and oxidative stress in neurodegenerative diseases, targeting mitochondrial pathways has emerged as a promising therapeutic strategy. Several approaches such as mitochondrial-targeted antioxidants, modulation of mitochondrial dynamics, and enhancement of bioenergetic function are currently being explored. Although challenges like BBB penetration persist, these interventions offer potential to slow or modify disease progression.
Oxidative stress, resulting from an imbalance between ROS and antioxidant defenses, is a key contributor to mitochondrial dysfunction. Mitochondrial-targeted antioxidants have shown encouraging results in preclinical models by preventing oxidative damage to neurons (Kumar, 2015). Compounds, such as MitoQ, MitoTEMPOL and mitovitE, are designed to mitigate oxidative stress at the source within mitochondria. However, their limited ability to cross the BBB remains a significant obstacle (Hara, 2019). Moreover, antioxidant therapy must be carefully regulated, as excessive antioxidant activity may disrupt essential oxidative signalling pathways required for normal cellular function (Hara, 2019).
The dynamic processes of mitochondrial fission and fusion are critical for maintaining mitochondrial health. Dysregulation of these processes has been implicated in multiple neurodegenerative diseases. Therapeutic targeting of proteins involved in mitochondrial dynamics, such as Drp1 or OPA1, has gained traction. For example, inhibition of Drp1 with the small molecule Mdivi-1 has been shown to restore mitochondrial morphology, improve dynamics, reduce amyloid pathology, and enhance cognitive performance in mouse models of early-stage AD, likely by reducing Aβ production (Wang, 2020).
Another emerging therapeutic strategy focuses on modulating mitochondrial bioenergetics. Enhancing mitochondrial function and ATP production may help compensate for the energy deficits commonly observed in neurodegenerative diseases. Oxaloacetate (OAA), a Krebs cycle intermediate, has demonstrated the ability to increase mitochondrial bioenergetic flux and improve brain function. While a phase I clinical trial in AD patients confirmed its safety, its cognitive benefits were limited (Swerdlow, 2016). Further studies are needed to determine effective dosing and long-term efficacy. Another candidate, CP2, a compound that accumulates selectively in neuronal mitochondria and inhibits mitochondrial complex I, has shown potential in AD mouse models by reducing amyloid plaques & tau pathology and preserving cognitive function (Zhang, 2015).
Peptide-based therapies, such as SS-31 (also known as elamipretide or MTP-131), represent another innovative approach. SS-31 targets the inner mitochondrial membrane and binds to cardiolipin, a lipid crucial for mitochondrial membrane integrity. This binding supports the formation of supercomplexes involved in OXPHOS. Preclinical studies have shown that SS-31 enhances mitochondrial and synaptic function in AD models, and it has recently been tested in clinical trials for rare diseases, such as Friedreich’s ataxia (NCT05168774).
In conclusion, therapeutic strategies aimed at correcting mitochondrial dysfunction hold significant promise for treating neurodegenerative diseases. However, several challenges remain, including optimizing drug delivery, ensuring BBB penetration, and maintaining a balance between ROS suppression and physiological signaling. Continued research is essential to refine these therapies and translate them into effective treatments for diseases such as AD, PD, and ALS.
Our team would be happy to answer any questions about mitochondrial dysfunction in microglia & astrocytes and neurodegenerative diseases or provide specific information about the AD, ALS, and PD models we use for therapeutic efficacy studies.
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on mitochondrial dysfunction in microglia & astrocytes and neurodegenerative diseases and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
Microglia-Neuron Interactions & Neurodegenerative Diseases
A concise review of the direct interactions between microglia & neurons, and how these cell-to-cell interactions may be affected in neurodegenerative diseases.
Microglia, Astrocytes & α-Synuclein in Parkinson’s Disease
How α-synuclein influences microglia and astrocytes in Parkinson’s disease and other synucleinopathies.
Lysosome Dysfunction in Microglia & Astrocytes
An overview of lysosomal dysfunction in microglia & astrocytes, and its role in neurodegenerative diseases.
TNF-α (TNF-alpha) & Microglia in Neurodegenerative Diseases
An overview of the function of tumor necrosis factor-alpha (TNF-α) in microglia and its contribution to the progression of neurodegeneration.
Astrocytes & Amyloid-β Mouse Models of Alzheimer's Disease
Analysis of astrocyte morphology in the amyloid-β plaque microenvironment provides a sensitive measure of disease progression in transgenic mice.
Mitochondrial Dysfunction & Parkinson's Disease
An overview of how mitochondrial dysfunction is associated with neurodegeneration in Parkinson’s disease.
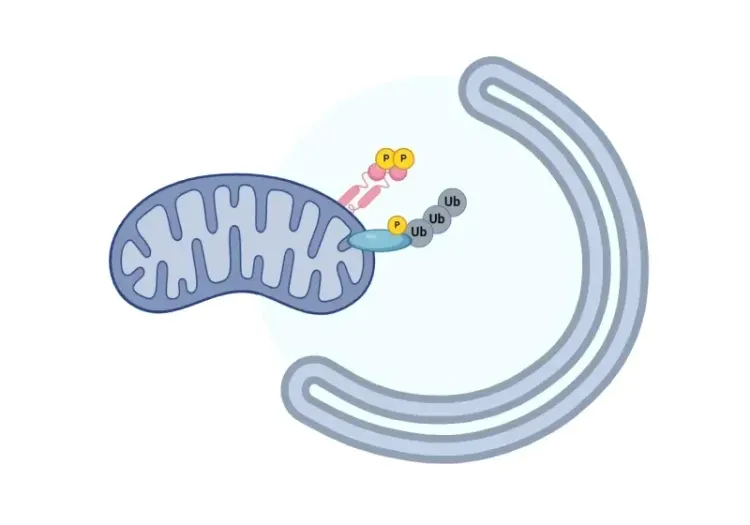
Mitophagy and Parkinson’s Disease
An overview of how impaired mitophagy can lead to neurodegeneration in Parkinson’s disease.