TNF-α 및 신경퇴행성 질환에서의 아스트로사이트
아스트로사이트에서의 TNF-α 신호전달 개요, 신경퇴행에서의 역할 및 이 경로를 표적으로 하는 치료 전략.
TNF-α 신호전달은 신경퇴행성 질환에서 성상세포 기능에 어떻게 영향을 미치는가?
아스트로사이트는 종양괴사인자-알파(TNF-α)에 대해 TNFR1과 TNFR2라는 두 가지 수용체 시스템을 통해 반응하며, 이들의 상반된 작용은 신경독성과 회복 사이의 균형을 유지한다. 실험적 자가면역 뇌척수염(EAE) 모델에서 보여지듯이, 아스트로사이트에서 TNFR1 활성화는 생체 내에서 시냅스 기능 장애와 기억력 결손을 유발하기에 충분하며, 해마 기능 장애는 글리아 TNF 신호 전달과 직접적으로 연관되어 있다(Habbas, 2015 ). 만성 예측 불가능한 경미한 스트레스(CUMS) 마우스와 같은 전임상 만성 스트레스 모델에서, TNFR1의 상향 조절은 성상세포증 및 신경세포 사멸로 특징지어지는 우울증 유사 증상과 연관되어 있는 반면, 약리학적 또는 유전적으로 TNFR1을 차단하면 이러한 효과가 역전됩니다(Gao, 2024 ). 반면, 아스트로사이트에서의 TNFR2 신호전달은 탈수초화 상태에서 염증 촉진 프로그램을 억제함으로써 반응성 아스트로글리오시스를 제한하고 재수초화를 촉진하는 동시에, 생리적 상태에서는 해마 시냅스 기능, 가소성 및 인지 기능을 지원합니다(Raphael, 2019 ; Carney, 2025 ).
이러한 수용체 간 차이는 치료적 기회를 창출한다:
- 주로 TNFR1을 활성화하는용해성 TNF(sTNF)의 선택적 중화는 탈수초화 모델(예: 큐프리존 탈수초화 모델)에서 재수초화를 향상시키면서, 막횡단 TNF(tmTNF)-TNFR2 경로를 통한 유익한 신호전달은 유지합니다(Karamita, 2017 ).
- 비선택적 TNF 차단의 합병증을 피하면서 이러한 차이를 활용하기 위해선택적 TNFR2 작용제가 활발히 개발 중입니다(Pegoretti, 2023 ).
이러한 연구 결과들은 TNFR1과 TNFR2가 신경교세포 내에서 신경독성과 회복 프로그램 사이의 균형을 조절하는 분자적 스위치 역할을 하며, 수용체 선택적 표적화가 유망한 치료적 접근법임을 강조합니다.
수용체 특이적 효과 외에도, TNF-α는 보호적 프로그램과 신경독성 프로그램 사이에서 신경교 세포 환경을 전환시키는 아스트로사이트 상태 전이를 조율합니다. 대표적인 예는 IL-1α, TNF-α, C1q로 구성된 사이토카인 삼중체로, 이는 항상성 아스트로사이트를 보체 C3 발현이 특징인 신경독성 A1 상태로 전환시킵니다. 이 경로의 차단이 A1 전환을 방지하고 ALS 모델에서 생존 기간을 연장함으로써, 아스트로사이트 TNF 신호전달이 질병 진행과 직접 연결됨을 보여준다(Liddelow, 2017 ; Guttenplan, 2020 ). 그러나 이 표현형이 항상 유해한 것은 아니다. 프리온 질환에서는 C3+ 아스트로사이트 제거가 퇴행을 악화시켜 질병 및 맥락 특이적 역할을 강조한다(Hartmann, 2019 ). 인간 iPSC 유래 아스트로사이트는 이러한 역학을 재현하는데, TNF-α가 단독으로 또는 IL-1β와 함께 NF-κB 활성화를 유발하고 C3를 상향 조절하여 기능적으로 글루타메이트 제거를 손상시킵니다 (Hyvärinen, 2019 ). 최근의 다중 오믹 프로파일링에 따르면, 인간 AD 및 ALS 뇌 조직에서 A1-유사 성상 세포는 신경 세포 손실 부위 주변에 클러스터를 형성하여 병인적 잠재력을 강화합니다 (Escartin, 2021 ). 파킨슨병(PD)에서 TNF-α에 노출된 아스트로사이트와 α-시누클레인 섬유에 노출된 아스트로사이트는 서로 다른 반응 패턴을 보이지만, 둘 다 미토콘드리아 기능 장애로 수렴하며, 이는 질병 진행에 기여하는 공통의 최종 결과입니다 (Russ, 2021 ). 이러한 연구 결과들은 TNF-α가 아스트로사이트 상태 전환의 핵심 촉진제 역할을 하여, 맥락에 따라 질병에 영향을 미치는 신경독성 프로그램으로 아스트로사이트를 유도함을 보여줍니다.
미세아교세포에서 TNF-α의 기능과 신경퇴행성 질환에 대한 기여에 대한 리뷰는 다음을 참조하십시오: 신경퇴행성 질환에서 TNF-α 및 미세아교세포
아스트로사이트 표현형 형성을 넘어, TNF-α는 신경 및 혈관 건강에 중요한 핵심 아스트로사이트 기능을 직접적으로 변화시킵니다:
- TNF-α는 TNFR1/NF-κB 경로를 통해 EAAT2/GLT-1을 하향 조절하여 글루타메이트 흡수를 감소시킴으로써 운동 뉴런에 대한 흥분성 독성 스트레스를 증가시킵니다(Jiang, 2019 ).
- 인간 iPSC 유래 아스트로사이트에서 TNF-α 및 기타 전염증성 사이토카인은 글루타메이트 제거를 감소시키고 면역 반응성 표지자를 강화합니다 (Hyvärinen, 2019 ).
- TNF-α는 신경교 전달을 강화합니다. 소뇌 회로에서, Bergmann 신경교 세포의 TNFR1 활성화는 글루타메이트 방출을 증가시키고 mGluR1 의존적 메커니즘을 통해 신경 세포의 흥분성을 변화시킵니다 (Shim, 2018 ).
- 염증 상태에서 TNF-STAT3 축은 혈액-뇌 장벽(BBB)의 무결성을 손상시키고, 염증에 의해 유발된 PD 성상 세포는 미세혈관 형태 형성을 지원하지 못하며, 이러한 결함은 MEK1/2 억제에 의해 역전됩니다 (de Rus Jacquet, 2023 ).
이러한 연구 결과는 TNF-α가 염증 상태 변화를 유발할 뿐만 아니라 글루타메이트 항상성, 신경-아교세포 간 소통, 혈관 안정성에서 아교세포의 필수적인 역할을 저해하여 신경퇴행성 과정을 가속화함을 보여줍니다.
이러한 다양한 결과는 TNF 수용체 하류 신호전달 네트워크에 의해 설명되며, 이는 염증 및 대사 허브를 아스트로사이트 상태 조절과 연결한다:
- NF-κB 활성화(RelA/p65)는 염증 전사 프로그램을 유도하고 미토콘드리아 기능 장애에 기여하며, 이러한 생체 에너지 결핍은 인간 성상 세포 모델에서 확인되었습니다(Russ, 2021 ).
- MAPK 신호 전달 모듈은 TNF 경로와 더욱 교차합니다: ERK/MAPK 활성은 PD 아스트로사이트에서 BBB 지원 기능을 조절합니다.
- 미세아교세포 TNF 하류의 JNK 활성화는 생체 내에서 신경-아교세포 간 소통을 재구성하는 아교세포 CXCL1 생산을 유도한다(Zhang, 2022 ; de Rus Jacquet, 2023 ).
종합하면, NF-κB 및 MAPK 허브와 통합된 TNFR1과 TNFR2의 이분법은 TNF-α를 신경퇴행성 질환 전반에 걸쳐 신경독성 또는 회복 프로그램을 조율하는 성상세포 상태 전이의 마스터 조절자로 자리매김합니다(Raphael, 2019 ).
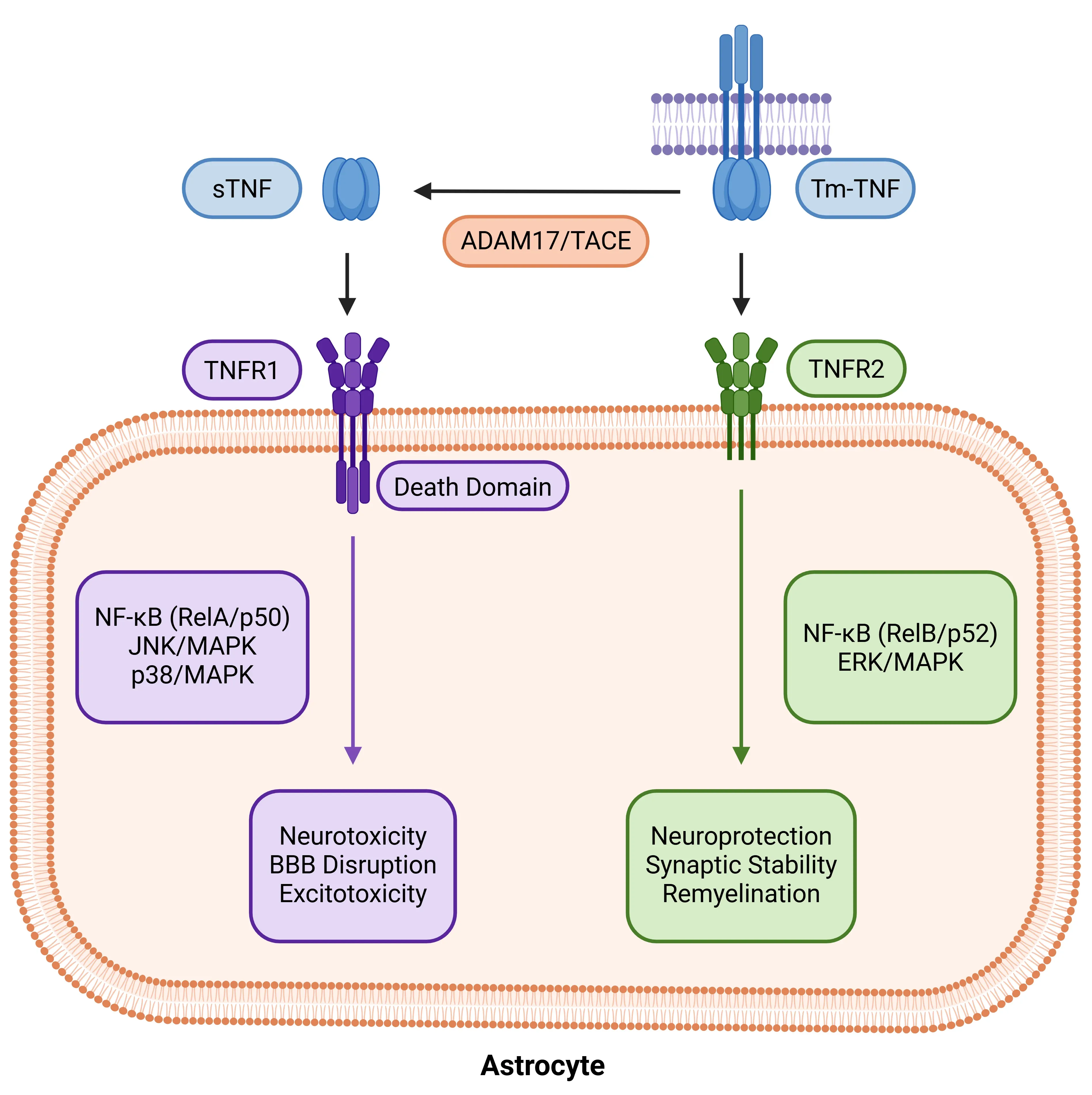
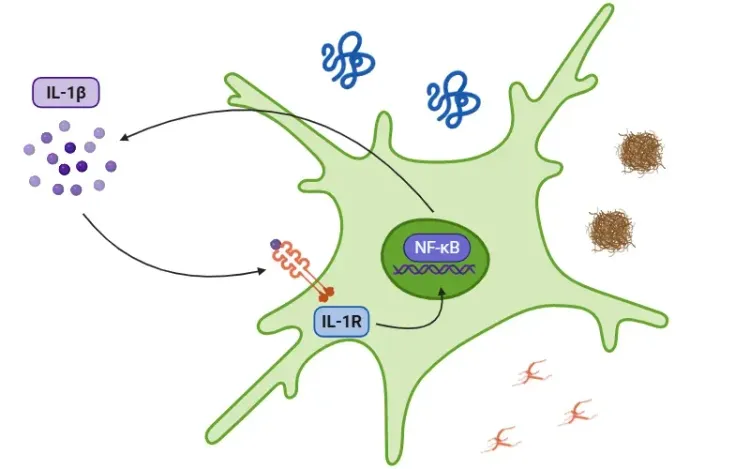
아스트로사이트에서의 TNF-α 수용체 이분화. 아스트로사이트의 TNF-α 반응은 리간드 형태와 수용체 특이성에 따라 달라진다. ADAM17/TACE에 의해 tmTNF에서 절단된 sTNF는 TNFR1을 활성화하여 NF-κB (RelA/p50), JNK 및 p38 MAPK 경로를 유발하며, 이는 신경독성과 혈뇌장벽(BBB) 파괴를 촉진한다. 반면, tmTNF는 TNFR2에 우선적으로 결합하여 비정형적 NF-κB(RelB/p52) 및 ERK/MAPK 경로를 통해 신경영양, 시냅스 안정성 및 재수초화를 촉진한다. 이러한 이분법적 특성은 TNF-α를 손상과 회복 사이의 아스트로사이트 상태를 조절하는 핵심 인자로 자리매김한다.
TNF-α 매개 신경염증에서 반응성 성상세포는 어떤 역할을 하는가?
TNF-α 환경 하의 반응성 성상세포는 단순한 신경교 활성화 이상의 다중 유해 역할을 수행한다. 본 연구에서는 이러한 상태가 다양한 질병 맥락에서 신경염증, 혈관 기능 장애 및 직접적인 신경세포 손상에 어떻게 기여하는지 설명한다.
아교세포 독성과 혈관 기능 장애
아스트로사이트의 반응성은 TNF-α에 의해 크게 형성되며, 이는 염증이 발생한 뇌에서 미세아교세포-아스트로사이트 간 소통의 핵심 증폭기 역할을 한다. 미세아교세포 유래 TNF-α는 IL-1α 및 C1q와 함께 체외 및 생체 내에서 항상성 아스트로사이트를 신경독성 A1 상태로 전환시키기에 충분하며, 이는 TNF-α가 아스트로글리오시스의 주요 유도인자임을 입증한다. 이 IL-1α/TNF/C1q 삼중체의 중화는 A1 전환을 방지하고 뉴런 및 올리고도교세포의 생존을 보존하여, TNF-α 의존적 반응성이 질병의 부산물이 아닌 병리의 원인임을 확인시켜 줍니다 (Liddelow, 2017 ). 해마 회로에서 TNF-α에 의한 아스트로사이트 TNFR1의 활성화는 흥분성 시냅스를 재구성하고 상황 기억을 손상시키는 아스트로사이트-뉴런 캐스케이드를 시작합니다 (Habbas, 2015 ).
혈관 인터페이스에서 TNF는 아스트로사이트를 STAT3 의존성 염증 반응 상태로 유도하여, 혈액-뇌 장벽(BBB)의 무결성을 파괴하고 혈관 내피 염증을 유발하는 인자를 분비하게 합니다(Kim, 2022 ). 이러한 혈관 장애 증거를 바탕으로, 최근 인간 뇌-칩 연구는 TNF에 의해 활성화된 아스트로사이트가 장벽 기능을 불안정하게 할 뿐만 아니라 미세혈관 형태 발생을 손상시키며, 이 결함은 MEK1/2 억제로 가역적임을 입증했습니다(de Rus Jacquet, 2023 ).
종합하면, TNF-α 반응성 아스트로사이트는 독성 글리아 상태를 촉진하고, 시냅스 통신을 방해하며, BBB 및 미세혈관 무결성을 약화시켜 신경염증성 손상을 증폭시킴으로써 기능 장애의 핵심 원인으로 작용한다.
신경염증의 염증성 및 산화적 유발인자
아스트로사이트가 TNF-α에 의해 유도된 반응성 상태를 취하면, 이들은 글리아 구획을 훨씬 넘어 손상을 확산시키는 염증 및 산화 스트레스의 활성 원천이 됩니다. 핵심 메커니즘은 NF-κB 경로의 활성화로, 이는 다음과 같은 전염증성 매개체의 생성을 유도합니다:
이러한 신호는 백혈구를 모집하고 중추신경계(CNS)의 국소 면역 활성을 증폭시킵니다(Giovannoni, 2020 ).
이와 병행하여 반응성 아스트로사이트는 활성산소종(ROS) 및 활성질소종(NOS)을 생성하여 TNF-α 신호전달을 산화 스트레스 경로와 연결시킵니다. 이는 신경 및 혈관 항상성을 더욱 불안정하게 만듭니다(Ding, 2021 ). 미세 아교세포와의 양방향 교신은 이러한 반응을 강화합니다: 성상 세포의 NF-κB 활성화는 미세 아교세포 증식과 백혈구 침윤을 유도하기에 충분하며, 이는 성상 세포가 수동적으로 반응하기보다 능동적으로 신경 염증을 확대한다는 것을 보여줍니다 (Ouali Alami, 2018 ).
참조: 미세아교세포 및 성상세포의 미토콘드리아 기능 장애
인간 iPSC 유래 아스트로사이트 모델은 이러한 특징을 재현하는데, IL-1β와 TNF-α의 복합 자극이 NF-κB 핵 전좌, 형태학적 재구성 및 면역 반응성 표현형 획득을 유도하기 때문입니다 (Hyvärinen, 2019 ). 이러한 실험 모델과 일관되게, 인간 AD 및 MS 조직의 단일 핵 RNA-seq (snRNA-seq)은 NF-κB에 의해 유도된 아스트로사이트 상태가 플라크와 병변 주변에 클러스터링되어 임상적 관련성이 직접적으로 있음을 보여줍니다 (Escartin, 2021 ).
이러한 연구 결과들은 TNF-α 반응성 아스트로사이트가 강력한 염증 및 산화 스트레스의 원동력으로 기능하며, 신경염증성 연쇄 반응을 적극적으로 증폭시키고 신경 및 혈관 취약성을 악화시킨다는 것을 보여줍니다.
신경독성과 회로 기능 장애
TNF-α에 의해 유도된 아스트로사이트 반응성은 정상적인 지지 역할을 박탈할 뿐만 아니라, 신경세포 및 올리고도교세포 손상의 능동적 원인이 됩니다. 기능적으로, A1 아스트로사이트는 정상적인 시냅스 형성 촉진 지원을 상실하고 신경세포 및 올리고도교세포를 사멸시키는 인자를 분비하여, TNF-α에 의해 유도된 반응 상태를 신경퇴화와 직접 연결합니다 (Liddelow, 2017 ). 한 가지 메커니즘은 APOE/APOJ 지질입자 내 포화 지질 분비를 포함한다; 지질 대사 효소 ELOVL1을 차단하면 이러한 독성을 방지할 수 있어, 아스트로사이트 지질 대사가 TNF 유발 신경세포 사멸과 연결됨을 시사한다 (Guttenplan, 2021 ).
회로 수준에서, 단독 또는 IL-6과 함께 지속된 TNF-α 노출은 인간 뉴런-아스트로사이트 공동 배양에서 뉴런 발화 패턴을 방해하여 염증 동안 인지 및 행동 장애의 원인이 될 수 있는 비정상적인 리듬을 초래합니다 (Goshi, 2025 ). 생체 내에서도 유사한 교란이 관찰되었는데, 만성적인 TNF-α 노출이 해마의 과흥분성과 간질형 활동을 유발하는 것으로 나타났습니다 (Vezzani, 2020 ).
전반적으로, TNF-α 반응성 아스트로사이트는 신경 세포 손실과 네트워크 불안정성의 중요한 원인으로 부상하며, 신경교 반응성을 신경 퇴행성 질환의 진행과 직접적으로 연결합니다.
이 모든 증거는 TNF-α가 아스트로사이트의 생물학적 특성을 근본적으로 재구성하여 중추신경계 항상성의 수호자에서 기능 장애의 핵심 원동력으로 전환시킨다는 것을 보여줍니다. TNF-α는 아스트로사이트를 염증, 혈관 손상, 신경 세포 취약성의 교차점에 위치시킴으로써, 이들을 수동적인 반응자가 아닌 신경 퇴행성 진행의 결정적인 조절자로 드러냅니다. 이러한 관점은 아스트로사이트를 배경적인 존재에서 만성 신경 염증과의 싸움에서 중추적인 치료 표적으로 재구성합니다.
TNF-α는 어떻게 아스트로사이트를 통해 신경퇴행성 질환 진행에 기여하는가?
만성적인 TNF-α 노출은 점차적으로 아스트로사이트를 유해한 상태로 몰아넣어 유전자 발현, 구조 및 대사를 변화시킵니다. 낮은 수준에서도 지속적인 TNF-α 신호는 아스트로사이트의 비대, 염증 유전자의 재구성, 대사 스트레스를 유발하여 연구자들이 아스트로사이트의 반응성을 단순히 "보호적이거나 독성"이 아닌 변화하는 상태의 스펙트럼으로 보게 합니다 (Escartin, 2021 ).
TNF-α를 포함하는 염증성 미세아교세포 신호는 신경독성 A1형 아스트로사이트로의 전환을 강화하여, 만성 선천성 면역 활성화와 신경퇴행성 질환에서 아스트로사이트 기능 장애 사이의 직접적인 연관성을 제시합니다 (Liddelow, 2017 ). 인간 iPSC 유래 성상 세포는 TNF-α (± IL-1β)가 NF-κB를 빠르게 활성화하고 형태 및 염증 유전자 네트워크를 재구성하는 이 과정을 재현합니다 (Hyvärinen, 2019 ). PD 관련 인간 성상 세포에서 TNF-α 또는 α-시누클레인 섬유는 미토콘드리아 호흡 결함을 특징으로 하는 중첩된 면역-대사 상태로 성상 세포를 유도하여, 장기간의 사이토카인 노출이 세포 에너지 균형을 어떻게 훼손하는지 보여줍니다 (Russ, 2021 ).
이러한 연구 결과들은 만성적인 TNF-α 신호전달이 아스트로사이트에 지속적인 염증 및 대사 부담을 각인시켜, 이들을 기능 장애 상태로 전환시킴으로써 지지 역할을 약화시키고 신경퇴행성 질환 진행에 적극적으로 기여함을 보여줍니다.
시냅스 수준에서 TNF-α는 아스트로사이트의 글루타메이트 처리 및 글리오트랜스미션을 변화시켜 신경 회로를 불안정하게 하고 흥분성 독성 취약성을 높인다:
- 아스트로사이트 TNFR1의 활성화만으로도 자가면역 탈수초화에서 흥분성 전달을 변화시키고 해마 기억을 손상시킬 수 있어, 네트워크 기능 장애에 아스트로사이트 TNF-α의 직접적인 인과적 역할을 입증합니다(Habbas, 2015 ).
- TNF-α는 TNFR1-NF-κB 경로를 통해 EAAT2/GLT-1 수송체 발현을 감소시켜 글루타메이트 흡수를 약화시키고 운동 뉴런을 독성 과자극에 노출시킵니다(Jiang, 2019 ).
- TNF-α와 IL-1β로 공동 자극된 인간 iPSC 유래 아스트로사이트는 글루타메이트 제거 장애와 더 강한 염증 활성화를 보입니다 (Hyvärinen, 2019 ).
- TNF-α는 Bergmann 신경교세포의 글루타메이트 방출을 증가시켜 소뇌 회로에서 신경교 전달을 강화하며, 이는 mGluR1 신호 전달을 통해 Purkinje 세포의 흥분성을 높이고 발화 패턴을 불안정하게 만듭니다 (Shim, 2018 ).
- 지속적인 TNF-α(±IL-6) 노출은 마이크로 전극 어레이(MEA) 상의 인간 뉴런-아스트로사이트 공동 배양에서 뉴런 발화 역학을 교란하여, 만성적인 사이토카인이 새로운 회로 부정맥을 유발함을 시사합니다(Goshi, 2025 ).
- 생체 내 연구에 따르면 TNF-α 과발현은 발작 감수성을 증가시키고 인지 기능 저하를 가속화하여 성상 세포 기능 장애를 광범위한 네트워크 불안정성과 연결합니다 (Vezzani, 2020 ).
이러한 연구 결과들은 TNF-α에 의해 유발된 아스트로사이트 기능 장애가 글루타메이트 조절과 시냅스 균형을 저해하여 광범위한 회로 불안정성과 진행성 인지 장애를 초래함을 입증합니다.
신경혈관 단위(NVU) 내에서 TNF-α에 의해 자극된 아스트로사이트는 염증 신호를 전달하여 혈액-뇌 장벽(BBB)의 무결성을 약화시키고 혈관 기능을 손상시킵니다. 인간 및 생체 외 모델에서 TNF-α는 아스트로사이트를 STAT3 의존적 반응 상태로 유도하며, 이는 SERPINA3(α1-항키모트립신, 급성기 세린 프로테아제 억제제)의 상향 조절로 특징지어지며, 이는 BBB 무결성을 감소시키고 내피 기능 장애를 촉진합니다(Kim, 2022 ).
PD 관련 LRRK2 돌연변이를 가진 아스트로사이트는 인간 뇌 칩 BBB 시스템에서 전염증성 프로파일을 채택하고 미세혈관 형태 형성을 지원하지 못합니다. 이러한 결함은 MEK1/2 억제에 의해 역전됩니다 (de Rus Jacquet, 2023 ). 이러한 발견은 아스트로사이트 말단발과 그 분비 분자가 BBB 안정성을 조절하며, 이러한 경계면의 염증성 재구성이 신경퇴행성 질환에서 혈관 기능 장애에 기여한다는 광범위한 증거와 일치한다(Yue, 2023 ). 이러한 메커니즘적 통찰과 일관되게, 인간 다발성 경화증 병변의 공간적 전사체학 분석은 TNF에 의해 활성화된 아스트로사이트가 혈관 주위 영역에 집적되어 사이토카인 풍부한 틈새를 생성함으로써 BBB 구조를 능동적으로 불안정화시킨다는 것을 보여줍니다(Mazziotti, 2024 ).
이러한 연구 결과는 TNF-α에 의해 유도된 아스트로사이트가 BBB의 무결성과 혈관 건강을 적극적으로 파괴하여 신경 퇴행성 질환에서 신경혈관 기능 장애의 주요 원인이 된다는 것을 보여줍니다.
주요 신경퇴행성 질환 전반에 걸쳐, TNF-α에 의해 유도된 성상세포 프로그램은 질병 진행의 공통적인 증폭기 역할을 합니다. 염증 신호 전달을 시냅스, 대사 및 혈관 기능 장애와 결합함으로써, 성상세포는 TNF-α 신호를 국소적 메커니즘으로 전환하여 신경세포 취약성과 조직 퇴화를 가속화합니다. 이러한 핵심 과정은 여러 질환에 공통적으로 나타나지만, TNF-α에 반응하는 성상세포가 질환별 특이적 환경과 상호작용하는 방식은 상당히 다양합니다. 다음 섹션에서는 알츠하이머병, 파킨슨병, ALS 및 다발성 경화증(MS)에서 성상세포를 통한 TNF-α 신호전달이 어떻게 진행에 기여하는지 설명합니다.
알츠하이머병(AD)
TNF-α는 아밀로이드-β 병리, 신경 염증 및 시냅스 기능 장애를 연결하는 AD의 핵심 매개체로 부상했습니다. 경증인지장애(MCI) 및 AD 환자의 뇌척수액(CSF)과 혈청 모두에서 TNF-α 수치가 지속적으로 상승한 것으로 보고되었으며, 기준선 농도가 높을수록 인지 기능 저하 속도가 빨라지고 치매로 진행될 위험이 높아지는 것으로 나타났습니다(Kim, 2017 ; Lista, 2024 ). 중요한 것은 이러한 생화학적 변화와 함께 아스트로사이트의 변화도 병행된다는 점입니다. 반응성 아스트로사이트 바이오마커(CSF GFAP 및 YKL-40 포함)는 아밀로이드-β와 타우가 해마 위축 및 인지 기능에 미치는 효과를 매개하며, 이는 살아있는 인간에서 아스트로사이트 반응성과 하류 신경퇴행 사이의 연관성을 보여줍니다(Ferrari-Souza, 2022 ; Pelkmans, 2024 ). 신경병리학 연구에 따르면 반응성 성상 세포는 베타 플라크와 신경섬유다발 주위에 군집하며 플라크 코어를 관통하여 병리의 억제 및 확산에 기여하는 것으로 밝혀졌습니다 (Perez-Nievas, 2018 ).
아밀로이드 베타 플라크 미세환경에서의 아스트로사이트 형태 분석은 다음을 참조하십시오: 아스트로사이트 및 아밀로이드 베타 알츠하이머병 마우스 모델
TNF-α가 알츠하이머병에 기여하는 핵심 메커니즘은 아스트로사이트에서 아밀로이드 생성 과정을 촉진하는 능력과 관련이 있습니다. 구체적으로, TNF-α 신호전달은:
- 아밀로이드 전구체 단백질(APP) 및 BACE1 발현을 상향 조절하여 Aβ 생성과 플라크 축적을 가속화합니다(Zhao, 2011 ).
- 흥분성-억제성 균형을 변화시켜 시냅스 항상성을 교란합니다
- 글루타메이트 전달을 강화
- GABA성 억제를 감소시킴
- AMPA 수용체 표면 이동 증가,
이러한 효과들은 종합적으로 해마 회로가 흥분성 독성에 취약해지도록 합니다 (Pribiag, 2013 ; Heir, 2020 ). 이 흥분성 독성 손상은 아스트로사이트 매개 글루타메이트 방출에 의해 더욱 증폭되며, 이는 신경세포 생존을 저해합니다 (Santello, 2011 ).
아스트로사이트 자체는 수동적 반응자가 아니라 알츠하이머병에서 TNF-α 신호전달의 핵심 촉진제이다. 실험적 증거는 다음과 같다:
- 미세아교세포가 결핍된 해마 배양에서는 TNF-α 신호전달이 지속되지만, 아스트로사이트 특이적 TNF 결손 시 소멸되어 활동 의존적 TNF 방출에 아스트로사이트가 필수적임을 입증함(Heir, 2024 ).
- 아스트로사이트의 NF-κB 활성화는 글루타메이트 유출에 대한 반응으로 TNF 생산을 조절하며, 사이토카인 주도 회로 재구성의 중심에 아스트로사이트를 위치시킵니다.
- 아스트로사이트 NF-κB 신호전달의 조절은 AD 모델에서 신경세포 및 시냅스 결과를 변화시킵니다(Jong Huat, 2024 ).
- 아스트로사이트의 글루타메이트 처리 장애, 특히 EAAT2/GLT-1 수송체 기능 저하는 TNF에 의한 흥분성 독성과 함께 신경 퇴행을 악화시킵니다 (Wood, 2022 ).
유전학 연구는 TNF-α의 병인적 역할을 강화합니다. TNF-α 프로모터의 G-308A 다형성은 전사 활성과 단백질 발현을 증가시키며, 여러 메타분석에서 이를 AD 감수성 증가와 연관시킵니다 (Wang, 2015 ). 이 변이는 APOE-ε4 유전자형과 시너지 효과를 발휘하여 염증 유발 및 질병 진행을 가속화할 수 있다(Contreras, 2020 ). 이는 산발성 및 가족성 알츠하이머병 전반에 걸쳐 공통 분모로서 TNF 신호전달의 중요성을 강조한다.
치료 연구는 TNF-α 표적화의 전환 가능성을 부각시킵니다. 데이터는 다음과 같습니다:
- 에타너셉트(Etanercept)나 아달리무맙(Adalimumab)과 같은 TNF 차단제로 치료받은 전신성 염증 질환 환자들은 치료받지 않은 집단에 비해 치매 발생률이 감소했습니다.
- 척추 주위 에타너셉트 투여를 이용한 소규모 파일럿 시험 및 사례 보고에 따르면, AD 환자에서 신속하고 때로는 지속적인 인지 기능 개선이 보고되었습니다.
- 피하 에타너셉트를 사용한 대규모 무작위 대조 시험에서는 큰 생물학적 제제의 혈뇌장벽 투과성 부족으로 인해 유의미한 이점이 입증되지 못했습니다.
- 혈뇌장벽 투과성 TNF 억제제를 사용한 전임상 연구에서는 AD 마우스 모델에서 인지 기능 개선과 함께 Aβ 및 타우 병리학이 감소하는 것으로 나타나, TNF 표적 치료의 근거를 강화한다(Plantone, 2024 ).
종합하면, 이러한 연구 결과들은 TNF-α가 아스트로사이트에 의한 기전을 통해 AD에서 아밀로이드 베타 축적, 흥분성 독성 및 신경퇴화의 핵심적인 원동력임을 입증하여 생물학적으로 타당한 치료 표적이 됨을 보여줍니다. 임상 데이터는 특히 혈뇌장벽 전달과 관련하여 아직 예비적 단계이지만, TNF-α 억제는 질병 수정을 위한 유망한 접근법을 제시합니다.
파킨슨병(PD)
성상세포 TNF-α는 파킨슨병(PD)에서 핵심적인 역할을 하며, α-시누클레인 병리와 신경염증 증폭 및 신경세포 취약성을 연결하는 매개체로 부상하고 있습니다. PD 환자에서 TNF-α 및 가용성 수용체는 뇌, 뇌척수액 및 혈액에서 지속적으로 상승되어 도파민 신경 퇴행, 인지 기능 저하 및 전반적인 질병 중증도와 상관 관계가 있습니다 (Liu, 2022 ). 아스트로사이트 내 알파-시누클레인의 축적은 미토콘드리아 및 소포체 기능을 방해할 뿐만 아니라 반응성, 친염증성 표현형을 유도합니다 (Wang, 2021 ). 중요한 것은, α-시누클레인 봉입체를 포함하는 성상 세포가 높은 수준의 TNF-α를 분비하여, 이 사이토카인을 단백질 병증과 염증 증폭을 연결하는 중심적인 효과기로 자리매김한다는 점입니다 (Lee, 2010 ).
아스트로사이트 유래 TNF-α는 파킨슨병에서 신경교 반응성의 피드 포워드 루프에 기여한다.
알파-시누클레인이 파킨슨병 및 기타 시누클레인병에서 미세아교세포와 성상세포에 미치는 영향에 대한 심층적인 검토는 다음을 참조하십시오: 파킨슨병에서 미세아교세포, 성상세포 및 알파-시누클레인
IL-1β 및 IL-6과 함께, 성상세포 TNF-α는:
- 미세아교세포 활성화를 증폭시켜 도파민 신경세포 손실을 가속화합니다.
- 신경세포 취약성을 악화시킵니다.
- 아스트로사이트에 의한 사이토카인 신호 전달을 진행성 흑질-선조체 퇴행과 연결하는 신경교 반응성의 피드포워드 루프를 형성합니다 (Wang, 2023 ).
실험적 PD 모델은 명백한 신경 세포 손실이 발생하기 전에도 미세 아교 세포와 성상 세포의 활성화를 유지하기 위해서는 IFN-γ와 TNF-α가 모두 필요함을 보여 주며, 이는 글리아에 의한 신경 염증을 시작하는 데 있어 TNF-α의 역할을 강조합니다 (Barcia, 2011 ). 기전 연구에 따르면 조절 인자 RGS5는:
- 성상세포에서 TNFR2 신호전달을 보호적에서 염증 촉진적 방향으로 전환시킴을 보여준다.
- TNFR1 활성화를 증폭시킵니다.
- 알파-시누클레인 응집, 신경퇴행 및 파킨슨병 모델에서의 사망률을 증가시킨다(Yin, 2023 ).
아스트로사이트가 신경독성 A1 상태로 전환되는 것은 TNF-α가 필수적인 또 다른 병리학적 기전이다. 미세아교세포가 분비하는 IL-1α, TNF-α, C1q가 함께 작용하여 아스트로사이트를 이 유해한 표현형으로 전환시킨다. A1 아스트로사이트는 파킨슨병 환자의 사후 뇌에서 확인되었다(Liddelow, 2017 ). 파킨슨병 동물 모델 (산발성 파킨슨병의 α-syn PFF 모델 및 hA53T Tg 마우스 모델)에서 병리적 α-시누클레인( ) 역시 A1 아스트로사이트 형성을 촉진하며, 이 과정을 차단하면 도파민성 뉴런을 보호하고 운동 기능을 보존한다(Yun, 2018 ). 이러한 A1 아스트로사이트는:
- 영양 및 시냅스 지원 능력을 상실합니다.
- 보체 성분 및 활성산소(ROS)를 포함한 신경독성 인자를 분비합니다.
- 신경 세포 손상을 악화시킵니다(Liddelow, 2017 ).
인간 연구는 파킨슨병(PD)에서 아스트로사이트 TNF-α의 병인적 역할을 확인한다. 인간 PD 중뇌의 단일 핵 RNA-seq 분석은 흑질 미세환경의 만성 염증 상태와 일치하는CD44high 아스트로사이트 표현형 및 사이토카인 신호 경로를 동반한 범-아교세포 활성화를 보여준다(Smajić, 2022 ). 배양된 인간 성상세포에서 TNF-α와 α-신경원섬유소 섬유는 미토콘드리아 호흡 장애를 동반한 면역 반응성 상태를 유발하며, 이는 파킨슨병 발병 기전과 일치하는 TNF 반응성 대사 취약성을 강조한다(Russ, 2021 ). 이 병리적 연쇄 반응은 보체 C4에 의해 더욱 증폭되며, 이는:
- -α-시누클레인에 대한 아스트로사이트의 전염증 반응을 증폭시킵니다.
- 신경세포 사멸 및 시냅스 병리를 촉진한다(Zou, 2025 ).
또한 염증에 노출된 인간 아스트로사이트는 뇌-칩 모델에서 미세혈관 형태 발생을 저해하고 혈뇌장벽(BBB) 무결성을 파괴한다(de Rus Jacquet, 2023). 기전적으로 TNF는 아스트로사이트를 STAT3 의존적 급성기 유사 상태로 유도하여 전신 염증을 뇌 내피세포로 전달함으로써 파킨슨병에서 BBB 파괴를 촉진한다(Kim, 2022 ).
종합하면, 이러한 연구 결과들은 아스트로사이트 TNF-α가 파킨슨병에서 α-시누클레인 병리, 신경교 증폭, 시냅스 기능 장애 및 혈관 손상을 연결하는 핵심 메커니즘임을 시사한다.
근위축성 측삭 경화증(ALS)
아스트로사이트 TNF-α 신호전달은 ALS에서 운동 뉴런 취약성의 초기적이고 지속적인 유발 요인으로 부상했다. ALS 환자의 뇌척수액(CSF)과 혈액에서 TNF-α 수치가 지속적으로 증가되어 질병 진행과 상관관계를 보인다(Jiang, 2022 ). 마찬가지로SOD1G93A 마우스에서는 증상 발현 이전에도 척수에서 TNF-α 및 그 수용체가 상향 조절되어 이 경로가 초기 병인에 기여함을 시사한다(Brambilla, 2016 ). 이러한 모델에서 유래한 성상세포-운동뉴런 공동배양에서, 운동뉴런 내막 결합형 TNF-α는 증가하는 반면 TNFR2 수준은 감소하여 수용체 신호전달의 불균형을 초래한다. 주목할 점은:
- TNFR1이 아닌 TNFR2를 삭제하면 운동 뉴런 생존이 회복되어, mTNF-α-TNFR2 축이 신경 독성의 핵심 매개체임을 확인하였다(Tortarolo, 2015 ).
- 반대로, TNFR1 신호전달은 아스트로사이트에서 글리아유래신경영양인자(GDNF) 방출을 통해 보호 효과를 발휘할 수 있는데, 이는 TNFR1 제거 시 결과가 악화되기 때문이다(Brambilla, 2016 ).
이러한 결과는 TNFR1과 TNFR2가 ALS 병리에 기여하는 방식이 고립된 공배양 시스템과 복잡한 생체 내 환경 사이에서 크게 달라지는 등 맥락에 매우 의존적임을 강조한다. 또한 TNFR1과 TNFR2 모두 운동 뉴런 사멸에 결정적으로 관여하는 세포사멸 신호 조절 키나아제 1(ASK1)/p38MAPK 경로를 활성화한다. P38 MAPK 억제는SOD1G93A 마우스 유래 성상세포/운동뉴런 공배양 모델에서 운동뉴런을 보호하며, 이 경로가 ALS에서 TNF 신호전달의 핵심 하류 효과기임을 부각시킵니다(Dewil, 2007 ). 동시에:
- 돌연변이 FUS를 보유한 성상세포에서 TNF-α는 NF-κB를 활성화하고 GluA2 통합을 감소시켜 AMPA 수용체 서브유닛 구성을 변화시킵니다.
- 이러한 변화는 운동 뉴런에서 칼슘 투과성과 흥분성 독성 스트레스를 증가시킵니다 (Kia, 2018 ).
- FUS-ALS 모델에서 TNF-α의 유전적 결손 또는 약리학적 중화는 운동 행동과 뉴런 생존을 회복시켜, 아스트로사이트 유래 TNF-α가 근접 독성 신호임을 확인했습니다(Jensen, 2022 ).
성상 세포 TNF-α는 또한 염증 환경을 형성하여 ALS 병리를 악화시키는 신경교 및 면역 반응을 모두 유도합니다. 주요 메커니즘은 다음과 같습니다.
- 아스트로사이트에서 유도 가능한 NF-κB 활성화는 미세아교세포 증식과 말초 백혈구 유입을 촉진하여 증상 진행을 가속화한다(Ouali Alami, 2018 ).
- 미세아교세포의 IL-1α/TNF/C1q 신호 삼중체는 신경독성 A1 아스트로사이트 표현형을 유도하며, 이 축을 차단하면SOD1G93A 마우스의 생존 기간이 연장됩니다(Guttenplan, 2020 ).
- SOD1 돌연변이를 가진 아스트로사이트는 미토콘드리아 스트레스, 프로테아좀 기능 장애 및 선천성 면역 활성화에 의해 유발되는 반응성, 기능 장애 상태를 취하며, 이는 TNF-α 신호 전달에 의해 악화됩니다.
- 섬유아세포 성장 인자 4(FGF4)는 일시적으로 성상 세포의 항상성을 회복시킬 수 있지만, TNF-α는 NF-κB 활성을 유지하여 이러한 보호 기능을 무효화하고 운동 뉴런을 취약한 상태로 남겨둔다(Velasquez, 2024 ).
아스트로사이트 TNF-α는 글루타메이트 항상성을 교란하여 ALS에서 흥분성 독성 스트레스를 더욱 강화합니다. 구체적으로, 아스트로사이트 TNFR1을 통한 TNF-α 신호 전달은
- EAAT2/GLT-1 발현을 억제합니다.
- 글루타메이트 방출을 촉진합니다.
- 글루타메이트 제거 장애와 운동 뉴런에 대한 흥분성 독성 스트레스를 유발하며, 이는 인간 ALS 병리와 일치하는 메커니즘입니다 (Jiang, 2019 ).
이러한 교란은 TNF에 의해 유발된 AMPA 수용체 구성의 변화와 시너지 효과를 발휘하여 칼슘에 의한 흥분성 독성을 강화합니다 (Kia, 2018 ; Jensen, 2022 ).
종합하면, 아스트로사이트 TNF-α는 ALS에서 염증, 수용체 불균형, 흥분독성을 연결하는 수렴점 역할을 합니다. 염증 캐스케이드를 증폭시키고, 글루타메이트 처리 방식을 변경하며, 흥분성 수용체 투과성을 동시에 증가시킴으로써 TNF-α는 운동 뉴런 퇴화를 가속화하고 질병 진행을 촉진합니다.
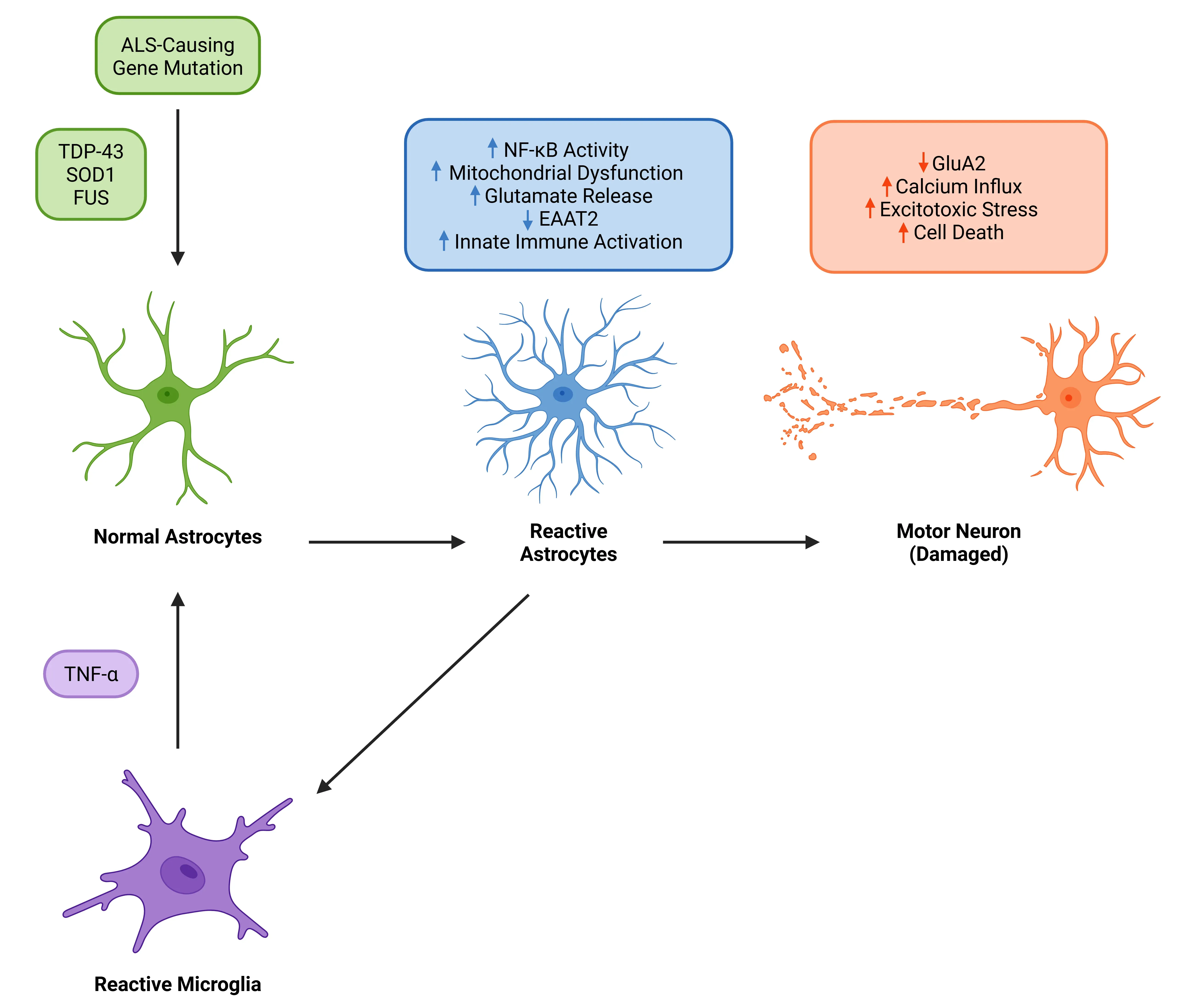
TNF-α는 ALS에서 성상세포 반응성과 운동뉴런 퇴화의 핵심 촉진제이다. TDP-43, SOD 또는 FUS 돌연변이와 연관된 ALS에서, 반응성 미세아교세포가 분비한 TNF-α는 TNFR 신호전달을 통해 성상세포 반응성을 촉진한다. 이는 아스트로사이트 내 NF-κB를 활성화하고 글루타메이트 제거를 저해하여 흥분성 독성 스트레스와 운동뉴런 세포 사멸을 초래한다. 반응성 아스트로사이트는 신경염증을 더욱 증폭시켜 운동뉴런 손실을 가속화하는 피드백 루프를 형성한다.
다발성 경화증(MS)
다발성 경화증(MS)에서 아스트로사이트는 TNF-α의 효과를 매개하는 핵심 요소로 부상하며, 염증 반응과 신경독성 및 회복 과정 모두를 연결합니다. TNF-α는 뇌척수액(CSF)에서 지속적으로 상승되어 있으며, 급성 및 만성 활성 MS 병변 모두에서 검출되며, 더 높은 수치는 더 심각한 장애 및 가속화된 질병 진행과 상관관계가 있습니다 (Sharief, 1991 ; Kosa, 2022 ). 병변 수준의 병리학은 급성 및 만성 활성 플라크에서 TNF의 상향 조절을 확인하여 TNF 신호 전달을 활성 조직 손상 부위와 일치시킵니다 (Mazziotti, 2024 ).
아스트로사이트가 국소 TNF 신호전달을 증폭시키는 핵심 기전은 막결합형 TNF의 단백질 분해적 가공과 관련됩니다. 활성 MS 병변 내에서 반응성 아스트로사이트는 ADAM17/TACE를 상향조절하여 tmTNF를 용해성 형태로 절단합니다. 이러한 전환은 TNFR1을 선택적으로 활성화하여 세포사멸 및 염증 촉진 캐스케이드를 강화합니다(Plumb, 2006 ). 실험적 자가면역 뇌척수염(EAE)에서, 아스트로사이트의 TNFR1 신호전달이 해마 시냅스 재구성과 인지 장애를 유발하는 것으로 밝혀져, 신경교 세포의 TNF 신호전달이 신경망 기능 장애와 직접 연결됨을 보여주었다(Habbas, 2015 ). 후속 유전자 구제 연구는 아스트로사이트에서만 선택적으로 TNFR1을 재활성화하는 것만으로도 시냅스 및 행동적 EAE 표현형을 재구축하기에 충분함을 입증하였다 (Di Castro, 2022 ). 종합하면, 이러한 발견들은 아스트로사이트를 단순한 반응자가 아닌 TNF 매개 신경독성의 증폭기로 자리매김한다.
동시에 아스트로사이트는 보호적 TNF 경로를 활용할 수 있어 다발성 경화증에서의 이중 역할을 강조한다. 이들은 TNFR1과 TNFR2를 모두 발현하며, TNFR2 신호전달이 신경보호 및 회복을 촉진한다는 증거가 축적되고 있다. 탈수초화 모델에서, 아스트로사이트 TNFR2 활성화는 항염증성 표현형을 촉진하고 pCXCL12에 의한 올리고도교세포 모집과 같은 메커니즘을 통해 재수초화를 지원합니다 (Brambilla, 2011 ; Patel, 2012 ). 최근 연구에 따르면 TNFR2는 아교세포의 친염증 프로그램을 억제하고 수초 형성에 대한 영양적 지원을 강화하여, TNFR1 매개 손상에 대한 균형 장치 역할을 한다는 점이 밝혀졌습니다(Raphael, 2019 ; Pegoretti, 2023 ).
이러한 기전적 통찰은 다발성 경화증(MS)에서 광범위한 항-TNF 치료의 임상적 시도가 실패한 이유를 설명하는 데 도움이 됩니다:
- 비선택적 TNF 차단은 다음과 연관되었습니다
- 탈수초화 사건의 역설적 악화
- 신경계 내 새로운 염증 증후군 발생 (W. Xie, 2024 )
대신 치료 방향은 TNF 신호전달의 선택적 조절로 전환되었습니다(Brambilla, 2011 ; Pegoretti, 2023 ):
- 전임상 및 중개 연구에서 유망한 전략은 다음과 같다
- TNFR1 억제
- STNF 중화
- TNFR2 보존 또는 작용
종합하면, 이러한 연구 결과들은 TNF가 다발성 경화증(MS)에서 아스트로사이트 반응을 조율하는 핵심 사이토카인으로 작용하며, 손상과 회복 사이의 균형을 지속적으로 기울인다는 점을 시사합니다. 아스트로사이트 TNF 신호전달의 이중적 특성은 치료적 표적화의 도전과 기회를 부각시키며, 수용체 선택적 조절을 신경보호 및 재수초화를 위한 매력적인 전략으로 만듭니다.
아스트로사이트에서 TNF-α 신호전달을 표적으로 하는 치료 전략은 무엇인가?
현재의 치료 전략은 아스트로사이트에서 TNF-α 신호전달의 서로 다른 측면을 표적으로 하는 네 가지 주요 접근법으로 분류할 수 있습니다:
TNF-α 수용체/리간드 억제제
치료 전략은 점차 병리적 TNFR1 활성을 억제하면서도 TNFR2의 재생 기능을 보존하거나 강화함으로써 아스트로사이트 내 TNF-α 신호전달을 정밀하게 조절하는 방향으로 나아가고 있습니다. 이 균형은 매우 중요합니다. 왜냐하면 TNFR1을 선택적으로 억제하거나 sTNF를 중화하면 중추 신경계 내에서 영양 및 재수초화 프로그램을 지원하는 tmTNF-TNFR2 신호 전달을 보존할 수 있기 때문입니다 (Fischer, 2020 ).
주요 접근법은 다음과 같다:
- 수용체 선택적 길항제
- 단가 인간 TNFR1 특이적 길항제인 아트로시맙(Atrosimab)은 EAE 모델을 포함한 염증 모델에서 효능을 입증하여, 선택적 TNFR1 차단을 아스트로사이트 중심의 관련 중재로 검증하였습니다.
- 추가적인 전임상 연구를 통해 이러한 이점이 급성 신경 퇴행성 질환으로까지 확대되어, 이 약물의 더 광범위한 신경 보호 잠재력을 강조하고 있습니다 (Ort-Casa, 2023 ).
- 리간드 표적 전략
- 지배적 음성 TNF 및 XPro1595(pegipanermin)는 mTNF-TNFR2 신호전달을 보존하면서 sTNF를 중화합니다(MacPherson, 2017 ; De Sousa Rodrigues, 2019 ).
- ADAM17/TACE 억제는 tmTNF가 sTNF로 절단되는 것을 방지하여 TNFR2 경로를 더욱 선호하게 합니다 (L. Xie, 2024 ).
이러한 유망한 결과에도 불구하고, 대부분의 생물학적 제제가 혈뇌장벽(BBB) 투과성이 낮아 임상 적용은 여전히 어려운 과제이다. 이러한 한계는 분자 셔틀 및 중추신경계(CNS) 표적 전달 시스템 개발을 촉진하였다(Kouhi, 2021 ). 또한 비선택적 전신성 TNF 억제제의 임상 경험은 탈수초화 질환의 역설적 악화를 보여주었으며, 이는 신경염증 환경에서 수용체 및 리간드 선택적 전략의 중요성을 강조한다(Mazziotti, 2024 ).
하류 신호 전달 표적화
또 다른 접근법은 TNF 수용체 하류 신호 전달 경로, 특히 아스트로사이트 반응성을 조절하는 NF-κB 및 MAPK 경로를 조절하는 것이다.
- 주요 하류 표적
- NF-κB: 아스트로사이트 특이적 활성화는 생체 내에서 신경염증을 증폭시키므로 선택적 억제가 유익할 수 있으나, 새로운 인간 세포 연구에 따르면 신경세포에 미치는 영향은 상황에 따라 보호적이거나 해로울 수 있음(Giovannoni, 2020 ).
- p38α MAPK: MW150과 같은 선택적 억제제는 AD 모델에서 신경염증을 억제하고 인지 기능을 개선하여 임상 적용 가능성을 시사합니다(Frazier, 2024 ).
- MEK1/2 (ERK 경로): 억제는 PD 관련 돌연변이를 가진 아스트로사이트에서 미세혈관 형태 형성을 회복시킵니다. 정상 조건에서 MEK/ERK 경로는 아스트로사이트가 스트레스 반응을 조절하고 균형 잡힌 염증 신호 전달을 구축하는 데 도움을 줍니다. 그러나 질병 상태에서는 이러한 보호 역할이 병리학적으로 증폭되어, MEK/ERK가 혈뇌장벽 안정화를 위한 조절 가능한 레버이자 신경퇴행성 질환에서 유망한 치료 표적임을 부각시킵니다(de Rus Jacquet, 2023 ).
- JNK: 억제는 성상 세포의 CXCL1 방출과 하류 신경 세포 스트레스를 제한하여 JNK를 병행 경로 표적으로 자리매김합니다 (Zhang, 2022 ).
아스트로사이트 표현형 조절
보완적 치료 전략은 아스트로사이트를 신경독성 표현형에서 벗어나 항상성 또는 회복 상태로 재프로그래밍하는 것을 목표로 합니다. IL-1α/TNF/C1q 축을 차단하면 A1형 아스트로사이트로의 전환을 방지하고 생체 내에서 신경세포 및 올리고도교세포의 생존력을 보존하여, 아스트로사이트 표현형 조절이 치료적 효과를 가질 수 있음을 가장 설득력 있게 입증한 사례 중 하나입니다(Liddelow, 2017 ). 다른 유망한 메커니즘으로는:
- ELOVL1 억제는 독성 지질 입자의 방출을 차단하여 지질 대사가 또 다른 약물 표적 취약점임을 강조합니다(Guttenplan, 2021 ).
- HDAC3 억제제는 염증 유전자 활성화를 감소시키면서 지지적 아스트로사이트 기능은 보존합니다(Clayton, 2024 ).
- GLP-1 수용체 작용제 (예: NLY01)는 미세아교세포에 의한 아스트로사이트 독성을 억제하며, 현재 알츠하이머병(AD) 및 파킨슨병(PD) 임상 시험이 진행 중입니다(Yun, 2018 ; Park, 2021 ).
유전자 치료 및 신개념 생물학적제제
유전자 및 생물학적 치료법은 아스트로사이트 특이적 TNF 신호 전달 조절 옵션을 더욱 확장하고 있습니다:
- 아스트로사이트에IL-2 유전자 전달은 전신 면역에 영향을 주지 않으면서 국소 조절 T 세포를 증가시키고 신경염증을 감소시킵니다(Yshii, 2022 ).
- GFAP 프로모터, 차세대 아스트로사이트 특이적 요소 및 광범위한 혈청형 증강제를 활용한 AAV 벡터의 발전으로, 현재 설치류와 대형 종 모두에서 아스트로사이트 선택적 유전자 전달이 가능해졌습니다(O’Carroll, 2021 ; Heffernan, 2022 ; Gleichman, 2023 ).
- TNFR2 작용제 투여 후 TNFR1 길항제 투여와 같은병용 전략은 인간화 EAE 모델에서 결과를 개선하며, 수용체 활성의 시간적 편향을 통해 위험을 최소화하면서 치료 효능을 극대화할 수 있음을 입증합니다(Pegoretti, 2023 ).
종합적으로, 아스트로사이트 TNF-α 신호전달을 표적화하는 것은 병리적 염증 반응을 억제하면서도 회복과 혈뇌장벽(BBB) 안정성을 지원하는 보호 기능을 보존할 필요성을 강조한다. 핵심 장벽은 여전히 기존 치료법의 제한된 뇌 투과성이지만, 선택적 억제제, 전달 시스템, 아스트로사이트 중심 중재법의 발전은 TNF-α 조절을 실행 가능한 신경보호 치료법으로 전환할 가능성을 제시한다.
TNF-α는 신경퇴행에서 다른 사이토카인과 어떻게 비교되나요?
신경퇴행성 질환에서 TNF-α는 다른 사이토카인과 몇 가지 핵심적인 차이점을 보이며, 이는 질병 진행에서의 역할과 치료 표적으로서의 잠재력을 모두 형성합니다.
TNF-α의 독특한 기능 (세포사멸, 괴사성 세포사멸, 염증)
TNF-α는 신경염증성 사이토카인 중 독특한 위치를 차지하는데, 그 수용체 연결이 염증 신호 전달을 프로그램된 세포사멸과 연결하기 때문입니다. 다른 매개체와 달리, TNF-α는 TNFR1을 통해 유전자 유도와 세포 사멸 및 괴사성 세포 사멸을 연결하고, NF-κB에 의한 생존 및 전염증성 산출물 외에도 RIPK1/RIPK3/MLKL 경로를 관여시킵니다 (Holbrook, 2019 ; van Loo, 2023 ).
반면:
- IL-6는 다음 경로를 통해 신호전달합니다
- 막 결합형 IL-6R을 통한 "전통적" 신호전달로, 보호 및 재생 반응을 촉진합니다.
- 용해성 IL-6R을 통한 "트랜스 신호전달"로 염증을 증폭시킵니다.
- 가용성 IL-6 수용체는 주로 작용제로 작용하며, 가용성 수용체가 길항제 역할을 하는 경향이 있는 TNF와는 대조적입니다 (Rose-John, 2021 ).
- STAT3 편향 반응을 활성화하고, 성상 세포 반응성을 증폭시키며, 혈관 안정성을 교란하여 혈액-뇌 장벽(BBB) 파괴에 기여합니다(Mora, 2024 ).
- 인터페론-감마(IFN-γ)는 아스트로사이트를 항원 제시 방향으로 재프로그래밍하여, TNF에 의해 유도된 세포사멸 상태 대신 인터페론 반응성 반응성 아스트로사이트(IRRA)를 생성합니다(Rostami, 2020 ; Prakash, 2024 ; Lee, 2023).
이러한 연구 결과는 TNF-α가 선천성 염증 신호와 사형 집행자 사멸 경로를 독특하게 연결하여, IL-6 또는 IFN-γ보다 신경 퇴행의 "최종 공통" 경로에 더 가깝게 위치한다는 것을 시사합니다.
아스트로사이트에서 TNF-α와 다른 사이토카인의 시너지 및 차이
기능적으로, TNF-α는 특히 다른 매개체와 함께 작용할 때, 아스트로사이트가 부적응 표현형을 준비하도록 하는 프라이밍 사이토카인 역할을 하는 경우가 많습니다.
주요 상호작용은 다음과 같습니다:
- TNF-α + IL-1α + C1q: 아스트로사이트를 A1-유사 상태로 유도하여 시냅스 지원 기능 상실 및 뉴런과 올리고도교세포의 사멸을 초래합니다 (Liddelow, 2017 ).
- TNF-α + IL-β: 인간 iPSC 유래 아스트로사이트에서 NF-κB 신호전달을 강력히 활성화시켜 구조를 재구성하고 강력한 면역 반응성 상태를 유도함(Hyvärinen, 2019 ).
이러한 연구 결과는 TNF-α가 IL-1α(C1q와 함께) 및 IL-1β와 시너지 효과를 발휘하여 신경독성 및 면역 반응성 아스트로사이트 상태를 유도하는 반면, IFN-γ 및 IL-6과 같은 사이토카인은 상이한 경로를 활성화하여 질병에서 사이토카인 프로그램이 아스트로사이트의 행동을 어떻게 형성하는지 강조한다는 점을 보여줍니다.
TNF에 의해 유도된 아스트로사이트의 공간적 및 단일 세포 서명
단일 세포 및 공간적 전사체 분석은 TNF-α에 반응하는 아스트로사이트가 질병과 관련된 독특한 상태를 형성함을 보여줍니다:
- 알츠하이머병( AD)에서
- snRNA-seq 분석에 따르면 플라크 인접 아스트로사이트는 정상적인 지지 유전자 프로그램을 상실하고 대신 TNF에 의해 유도된 보체 및 염증 경로를 활성화한다(Dai, 2023 ).
- 공간적 전사체학은 이러한 플라크 경계 아스트로사이트가 TNF/IL-1 신호에 의해 강력하게 형성되며 퇴화하는 뉴런 주변에 군집함을 추가로 확인한다(He, 2024 ).
- 메타분석에 따르면 알츠하이머병 진행 단계에 따라 TNF-α, IL-6, IL-1β가 병행 증가하며, 이는 인지 기능 저하 악화와 직접적인 상관관계를 보입니다(Lista, 2024 ; Serna, 2025 ).
- 파킨슨병( PD)에서
- snRNA-seq 분석은 흑질에서 만성적인 TNF/IL-1 유도 염증을 반영하는 유전자 발현을 보이는 CD44 고발현 아스트로사이트 하위 집단을 포함하여 광범위한 신경교세포 활성화가 존재함을 보여줍니다(Smajić, 2022 ).
TNF-α 대 기타 사이토카인의 치료적 선택성
TNF-α는 신호 전달 구조가 유해한 TNFR1 경로를 선택적으로 표적화하면서도 보호적 TNFR2 기능을 보존할 수 있어 치료적으로 두드러집니다. 이러한 선택적 균형은 TNF에 고유하며 IL-6 또는 IFN-γ 차단으로는 달성할 수 없습니다(Fischer, 2020 ; Papazian, 2021 ).
- 비선택적 항-TNF 치료법은 중추신경계 자가면역 반응을 악화시킬 수 있다(Mazziotti, 2024 ).
- IL-6 치료제 (예: sgp130Fc)는 보호적 "전통적" IL-6 신호전달은 유지한 채 염증을 차단하지만, TNF 수용체 신호전달에 고유한 세포사멸 경로는 해결하지 못합니다(Rose-John, 2021 ).
- IFN-γ 치료는 TNF에 의해 유도되는 세포 사멸 및 괴사성 사멸 경로보다는 항원 제시와 같은 면역 관련 성상 세포 기능을 조절합니다 (Prakash, 2024 ; Lee, 2023 ).
이러한 비교를 종합하면, TNF-α는 사이토카인 중에서도 신호전달을 치료적으로 정밀 조절할 수 있다는 점에서 독특합니다: TNFR1 또는 sTNF의 선택적 차단은 신경독성 및 세포사멸 경로를 억제하면서도 TNFR2 매개 회복 기능을 보존하는데, 이는 IL-6 또는 IFN-γ 개입으로는 달성할 수 없는 치료적 균형입니다.
종합적으로, TNF-α는 다른 사이토카인보다 신경퇴행성 진행과 더 직접적으로 연관된 방식으로 아스트로사이트의 행동을 형성한다는 증거가 제시된다. 염증 신호를 세포사멸 경로에 연결하고, 부적응적 아스트로사이트 상태를 증폭시키며, 영역 특이적 전사 변화를 유도함으로써, TNF-α는 아스트로사이트를 질병 메커니즘과 치료 기회의 중심에 위치시킨다.
저희 팀은 아스트로사이트에서 TNF-α의 기능 및 신경퇴행성 질환에 대한 기여도에 관한 질문에 기꺼이 답변해 드릴 수 있으며, 치료 효능 연구에 활용하는 알츠하이머병(AD), 근위축성 측삭경화증(ALS), 파킨슨병(PD) 모델에 대한 구체적인 정보를 제공해 드릴 수 있습니다.
신경퇴행성 질환 모델에 대해 자세히 알아보기
관련 콘텐츠
신경퇴행성 질환에서 TNF-α 및 미세아교세포에 관한 최신 정보와 신경퇴행성 질환 동물 모델에서 치료제 평가와 관련된 모범 사례.
신경 퇴행성 질환의 TNF-α & 미세 아교 세포
미세아교세포에서 종양괴사인자-알파(TNF-α)의 기능과 신경퇴행의 진행에 대한 기여에 대한 개요.
IL-1β란 무엇인가요?
IL-1β의 개요, 전신 및 신경계 질환에서의 염증 촉진 역할, 그리고 IL-1β 억제를 포함한 치료 전략.
미토콘드리아 기능 장애와 미세아교세포 및 별아교세포
미토콘드리아 기능 장애가 알츠하이머 병, 파킨슨 병, ALS를 포함한 신경퇴행성 질환에서 미세아교세포와 별아교세포에 미치는 역할.
미세아교세포, 아스트로사이트 및 타우 단백질의 신경퇴행성 질환에서의 역할
글리아 세포에 의해 유발된 신경염증이 알츠하이머 병 및 기타 타우 병증에서 타우 단백질의 응집, 확산, 및 신경세포 손실을 촉진하는 메커니즘.
인터루킨-1 베타 (IL-1β)와 신경퇴행성 질환
IL-1베타가 알츠하이머 병(AD), 파킨슨 병(PD), 및 근위축성 측삭경화증(ALS)을 포함한 신경퇴행성 질환에서의 역할.
미세아교세포, 아스트로사이트 및 타우 단백질의 신경퇴행성 질환에서의 역할
글리아 세포에 의해 유발된 신경염증이 알츠하이머 병 및 기타 타우 병증에서 타우 단백질의 응집, 확산, 및 신경세포 손실을 촉진하는 메커니즘.