APOE4, Microglia & Alzheimer’s Disease
An overview of how ApoE4 influences microglial activity in Alzheimer's disease and the development of targeted therapeutics.
What is APOE4?
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by the gradual decline in memory and cognition. AD is the most common cause of dementia in individuals over the age of 65, and its prevalence is rising globally due to the aging population. The disease is associated with the accumulation of two hallmark proteins in the brain: amyloid-β (Aβ) plaques and tau tangles. These deposits disrupt neuronal communication, leading to cell death and cognitive impairment.
AD is categorized into two forms:
- Early-onset Alzheimer’s disease (EOAD), which is rare and typically develops before the age of 65.
- Late-onset Alzheimer’s disease (LOAD), which accounts for over 95% of AD cases and typically occurs after age 65.
While the exact cause of AD remains unclear, it is understood that a combination of genetic, environmental, and lifestyle factors contribute to the disease. Of these, genetic predisposition is a significant risk factor, and one of the most well-known genetic contributors to AD is the APOE gene.
The APOE (apolipoprotein E) gene is located on chromosome 19 and encodes the apolipoprotein E (ApoE) protein, which plays a key role in lipid metabolism, neuronal repair, and immune function within the central nervous system (CNS). ApoE is involved in the transport of lipids, including cholesterol, which are crucial for neuronal integrity and synaptic function.
The APOE gene exists in three major isoforms – APOE ε2, APOE ε3, and APOE ε4 – which differ by a single amino acid substitution. APOE ε3 is the most common and considered to be “neutral” with respect to AD risk, while APOE ε2 is thought to be protective. However, APOE ε4 significantly increases the risk of developing LOAD.
The pathological mechanisms underlying this increased risk are complex and involve the interaction between ApoE4 and various cellular processes in the brain. Specifically, ApoE4 is believed to impair lipid metabolism, disrupt microglial function, and interfere with Aβ clearance, all of which contribute to the accumulation of toxic proteins and chronic inflammation in the brain.
In addition, ApoE4 promotes Aβ aggregation, facilitating the formation of plaques that damage neurons. ApoE4 also modulates tau pathology, promoting the hyperphosphorylation of tau, which leads to the formation of neurofibrillary tangles – a hallmark feature of AD. The combination of impaired clearance of Aβ, tau aggregation, and neuroinflammation drives the progression of the disease, leading to neuronal loss and cognitive decline (Shi, 2017; Najm, 2020).
How do APoE4-microglial interactions drive AD progression?
Microglia are the resident immune cells of the CNS, making up approximately 10-15% of the cells in brain. They play an essential role in maintaining brain homeostasis by constantly surveying the brain environment for damage, clearing debris, and regulating inflammation. Under normal conditions, microglia help protect neurons from injury and support synaptic function by clearing cellular waste, including misfolded proteins, such as Aβ and tau. However, in AD, particularly in individuals carrying the APOE ε4 allele, microglia undergo dysfunction, which contributes to the disease’s progression.
In the presence of ApoE4, microglial function is significantly altered in the following ways:
Impaired Clearance of Aβ
- ApoE4 reduces the ability of microglia to effectively clear Aβ peptides, leading to plaque accumulation (Deane, 2008; Najm, 2020).
- Normally, microglia express P2RY12 receptors, which are essential for guiding microglia to Aβ deposits and enabling their clearance. In ApoE4 carriers, P2RY12 receptor expression is downregulated, impairing microglial motility and their ability to effectively remove Aβ plaques (Sepulveda, 2024).
For an analysis of the complex spatial relationships between Aβ plaques, activated & resting microglia, and astrocytes in an APP/PS1 transgenic model, see: Amyloid-β & Inflammatory Microenvironment in Alzheimer's Mice
Pro-Inflammatory Environment
- The accumulation of Aβ triggers microglial activation, a process meant to promote the clearance of cellular debris and initiate immune responses. However, in the presence of ApoE4, microglia adopt a chronic pro-inflammatory phenotype that exacerbates neuroinflammation (Rao, 2025).
- Microglia expressing ApoE4 release high levels of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6 (Iannucci, 2021). These cytokines perpetuate chronic inflammation in the brain.
- ApoE4 also increases the expression of TREM2, a receptor involved in the activation of disease-associated microglial (DAM). While DAM activation is aimed at clearing Aβ, it further promotes inflammation and neurodegeneration (Iannucci, 2021).
For more information on the role of pro-inflammatory cytokine, IL-1β, in neurodegenerative diseases, see: Interleukin-1 Beta (IL-1β) and Neurodegenerative Diseases
Aβ and Tau Pathology
- ApoE4 promotes the formation of neurotoxic Aβ oligomers, worsening amyloid pathology (Hashimoto, 2012).
- In addition to Aβ, ApoE4 also increases tau hyperphosphorylation, which leads to the formation of neurofibrillary tangles, a hallmark of AD (Shi, 2017).
Disruption of Lipid Metabolism
- ApoE4 impairs lipid metabolism, leading to the accumulation of intracellular lipid droplets and cholesterol in microglia (Wang, 2022).
- This lipid accumulation compromises microglial function, further impairing the ability to clear Aβ and other toxic proteins.
- Moreover, these lipid imbalances promote pro-inflammatory signaling within microglia, further exacerbating neurodegeneration.
Increased Oxidative Stress
- ApoE4-expressing microglia produce elevated levels of reactive oxygen species (ROS), leading to oxidative stress in the brain (Liu, 2025).
- ROS damage cellular components, and oxidative stress further impairs microglial function, promoting a vicious cycle of inflammation and neurodegeneration.
See: Mitochondrial Dysfunction in Microglia & Astrocytes
Morphological Changes in Microglia
- Normally, microglia have extensive, highly ramified processes that enable them to efficiently survey the brain and respond to damage.
- ApoE4 alters the morphology of microglia, causing them to have shortened and less ramified processes, impairing their ability to survey the brain for damage and clear Aβ and tau (Sepulveda, 2024).
For an in-depth review of microglial morphological changes in neurodegenerative diseases, see: Microglia Morphology in ALS, Alzheimer's Disease & Parkinson's Disease
Together, these dysfunctional interactions between ApoE4 and microglia create a self-perpetuating cycle of neuroinflammation, amyloid plaque accumulation, tau pathology, and oxidative stress. These pathological events not only exacerbate neuronal damage, but also accelerate the progression of AD, leading to cognitive decline and neurodegeneration.
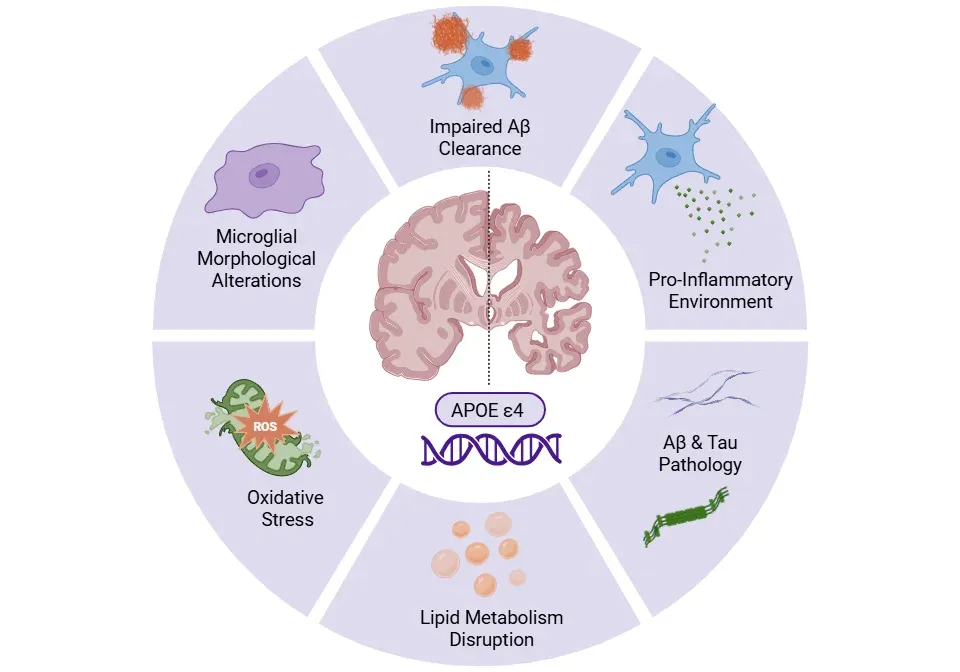
ApoE4-driven microglial dysfunction in Alzheimer's disease.
ApoE4 alters microglial morphology and function, impairs Aβ clearance, and disrupts lipid metabolism, promoting Aβ and tau pathology, a pro-inflammatory environment, and oxidative stress – collectively accelerating neurodegeneration.
What therapeutic strategies target ApoE4 in microglia?
Given the crucial role of ApoE4 in driving AD progression, particularly through its effects on microglial function and neuroinflammation, several therapeutic strategies have been developed to target ApoE4 directly or modulate its effects. These strategies aim to either correct ApoE4’s structure, reduce its expression, or counteract its downstream pathological effects.
Small Molecules
- Small molecules have been designed to specifically alter the structure and function of ApoE4, with the goal of stabilizing the protein structure and preventing its pathogenic conformational shifts (Dias, 2025).
- Phase III clinical trials of ALZ-801 (valiltramiprosate) in APOE ε4 homozygotes with mild cognitive impairment (MCI) have shown promising results (NCT04770220, NCT06304883).
Antisense Oligonucleotides (ASOs)
- ASOs are synthetic strands of oligodeoxynucleotides designed to bind to messenger RNA and reduce expression of a specific protein – in this case, ApoE4.
- Preclinical studies have demonstrated that ApoE4-targeting ASOs can effectively reduce ApoE4 expression and decrease Aβ pathology in mouse models of AD.
- Notably, when administered prior to plaque deposition, ASOs were able to lower Aβ load. However, when administered during the seeding stage, ASOs were found to lead to larger Aβ plaques, but with reduced plaque-associated neuritic dystrophy, thereby highlighting the timing-dependent effects of ASOs (Huynh, 2017).
- ASOs targeting ApoE4 have also been shown to reduce tau pathology, preserve synaptic density, and reduce neuroinflammation in tauopathy mouse models (Litvinchuk, 2021).
Gene Editing
- Gene editing technologies, such as CRISPR/Cas9, offer the potential to directly alter the APOE ε4 allele at the genetic level. By correcting the ε4 allele to the ε3 or ε2 isoforms, gene editing could potentially reverse the underlying genetic risk for AD.
- In vitro models using CRISPR/Cas9 have shown promising results in reducing tau phosphorylation, cytotoxicity, and Aβ pathology by improving the clearance of Aβ by glial cells (Raulin, 2022).
- AAV-based gene therapies, like LX1001, which uses adeno-associated virus (AAV) vectors to deliver the APOE2 gene into the brain, have shown early-phase clinical trial success in moderate AD and MCI. Interim data from these trials indicate increased ApoE2 expression, reduced tau biomarkers, and stabilized Aβ pathology (NCT03634007, NCT05400330).
Anti-Inflammatory Drugs (NSAIDs)
- Non-steroidal anti-inflammatory drugs (NSAIDs) are one of the most widely used classes of drugs to reduce microglial activation and cytokine release. However, clinical outcomes with NSAIDs have been variable and have not consistently shown efficacy in AD (McGeer, 2007; Imbimbo, 2010).
Immunotherapies
- Immunotherapies targeting ApoE4 represent another strategy for reducing neuroinflammation and mitigating the pathological effects of ApoE4 in AD.
- Preclinical studies in AD models have explored anti-ApoE antibodies to directly target and neutralize ApoE, thereby reducing its pathological effects. In these models, anti-ApoE antibodies have been shown to promote the recruitment of microglia around Aβ plaques and reduce amyloid burden (Kim, 2012; Liao, 2018).
These therapeutic strategies are still in various stages of development, but they represent promising approaches to mitigate the harmful effects of ApoE4 in AD. Future clinical trials will be crucial in determining the safety, efficacy, and long-term impact of these interventions.
Our team would be happy to answer any questions about how ApoE4 influences microglial activity in Alzheimer's disease or provide specific information about the models we use for therapeutic efficacy studies.
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on Neuroinflammation and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
TNF-α (TNF-alpha) & Microglia in Neurodegenerative Diseases
An overview of the function of tumor necrosis factor-alpha (TNF-α) in microglia and its contribution to the progression of neurodegeneration.
Mitochondrial Dysfunction in Microglia & Astrocytes
The role of mitochondrial dysfunction in microglia and astrocytes in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and ALS.
What Is IL-1β (IL-1b)? Function, Signaling, and Biological Role
An overview of IL-1β, including its signaling pathways, involvement in disease mechanisms, and potential therapeutic targets.
Interleukin-1 Beta (IL-1β) and Neurodegenerative Diseases
The role of IL-1beta in neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS).
Lysosome Dysfunction in Microglia & Astrocytes
An overview of lysosomal dysfunction in microglia & astrocytes, and its role in neurodegenerative diseases.