TNF-α (TNF-alpha) & Astrocytes in Neurodegenerative Diseases
An overview of TNF-α signaling in astrocytes, its role in neurodegeneration, and therapeutic strategies targeting this pathway.
This resource describes:
- How does TNF-α signaling affect astrocyte function in neurodegenerative diseases?
- What roles do reactive astrocytes play in TNF-α–mediated neuroinflammation?
- How does TNF-α contribute to neurodegenerative disease progression through astrocytes?
- What therapeutic strategies target TNF-α signaling in astrocytes?
- How does TNF-α compare to other cytokines in neurodegeneration?
How does TNF-α signaling affect astrocyte function in neurodegenerative diseases?
Astrocytes respond to tumor necrosis factor-α (TNF-α) through two receptor systems, TNFR1 and TNFR2, whose opposing activities establish the balance between neurotoxicity and repair. TNFR1 activation in astrocytes is sufficient to induce synaptic dysfunction and memory deficits in vivo, as shown in the experimental autoimmune encephalomyelitis (EAE) model, where hippocampal impairment is directly linked to glial TNF signaling (Habbas, 2015). In preclinical chronic stress models, such as chronic unpredictable mild stress (CUMS) mice, TNFR1 upregulation is linked to depression-like symptoms marked by astrogliosis and neuronal apoptosis, while blocking TNFR1, pharmacologically or genetically, reverses these effects (Gao, 2024). In contrast, TNFR2 signaling in astrocytes limits reactive astrogliosis and promotes remyelination by suppressing pro-inflammatory programs in demyelinating conditions, while also supporting hippocampal synaptic function, plasticity, and cognition under physiological states (Raphael, 2019; Carney, 2025).
This receptor divergence creates therapeutic opportunities:
- Selective neutralization of soluble TNF (sTNF), which mainly activates TNFR1, enhances remyelination in demyelination models (e.g. cuprizone demyelination model), while preserving beneficial signaling through the transmembrane TNF (tmTNF)–TNFR2 pathway (Karamita, 2017).
- Selective TNFR2 agonists are under active development to exploit this divergence while avoiding the complications of non-selective TNF blockade (Pegoretti, 2023).
Together, these findings highlight TNFR1 and TNFR2 as a molecular switch in astrocytes, tipping the balance between neurotoxic and repair programs, and underscore receptor-selective targeting as a promising therapeutic avenue.
Beyond receptor-specific effects, TNF-α orchestrates astrocytic state transitions that shift the glial landscape between protective and neurotoxic programs. A prominent example is the cytokine triad of IL-1α, TNF-α, and C1q, which drives homeostatic astrocytes into neurotoxic A1 states marked by complement C3 expression. Disruption of this pathway prevents A1 conversion and extends survival in ALS models, directly linking astrocytic TNF signaling to disease progression (Liddelow, 2017; Guttenplan, 2020). Yet, this phenotype is not universally harmful; in prion disease, ablation of C3+ astrocytes worsens degeneration, underscoring disease- and context-specific roles (Hartmann, 2019). Human iPSC-derived astrocytes replicate these dynamics, where TNF-α, alone or with IL-1β, triggers NF-κB activation and C3 upregulation, functionally impairing glutamate clearance (Hyvärinen, 2019). Recent multi-omic profiling shows that A1-like astrocytes in human AD and ALS brain tissues cluster around sites of neuronal loss, reinforcing their pathogenic potential (Escartin, 2021). In Parkinson’s disease (PD), astrocytes exposed to TNF-α versus α-synuclein fibrils follow different reactive patterns, yet both converge on mitochondrial dysfunction, a shared endpoint that contributes to disease progression (Russ, 2021). Together, these findings show that TNF-α is a key driver of astrocytic state transitions, pushing them toward neurotoxic programs that influence disease in context-dependent ways.
For a review on the function of TNF-α in microglia and its contribution to neurodegenerative diseases, see: TNF-α & Microglia in Neurodegenerative Diseases
Beyond shaping astrocytic phenotype, TNF-α directly alters key astrocytic functions that are critical for neuronal and vascular health:
- TNF-α reduces glutamate uptake by downregulating EAAT2/GLT-1 through a TNFR1/NF-κB pathway, thereby increasing excitotoxic stress on motor neurons (Jiang, 2019).
- In human iPSC-derived astrocytes, TNF-α and other pro-inflammatory cytokines diminish glutamate clearance and enhance immune-reactive signatures (Hyvärinen, 2019).
- TNF-α enhances gliotransmission. In cerebellar circuits, TNFR1 activation in Bergmann glia increases glutamate release and alters neuronal excitability through mGluR1-dependent mechanisms (Shim, 2018).
- A TNF-STAT3 axis under inflammatory conditions compromises blood-brain barrier (BBB) integrity, and inflammation-primed PD astrocytes fail to support microvascular morphogenesis - a defect reversed by MEK1/2 inhibition (de Rus Jacquet, 2023).
These findings demonstrate that TNF-α not only drives inflammatory state changes but also undermines essential astrocytic roles in glutamate homeostasis, neuron-glia communication, and vascular stability, thereby accelerating neurodegenerative processes.
These diverse outcomes are explained by the signaling networks downstream of TNF receptors, which couple inflammatory and metabolic hubs to astrocytic state control:
- NF-κB activation (RelA/p65) drives inflammatory transcriptional programs and contributes to mitochondrial dysfunction, with these bioenergetic deficits confirmed in human astrocyte models (Russ, 2021).
- MAPK signaling modules further intersect with TNF pathways: ERK/MAPK activity regulates BBB support functions in PD astrocytes.
- JNK activation downstream of microglial TNF induces astrocytic CXCL1 production that reshapes neuron-glia communication in vivo (Zhang, 2022; de Rus Jacquet, 2023).
Taken together, the dichotomy between TNFR1 and TNFR2, integrated with NF-κB and MAPK hubs, positions TNF-α as a master regulator of astrocyte state transitions, orchestrating either neurotoxic or reparative programs across neurodegenerative disease (Raphael, 2019).
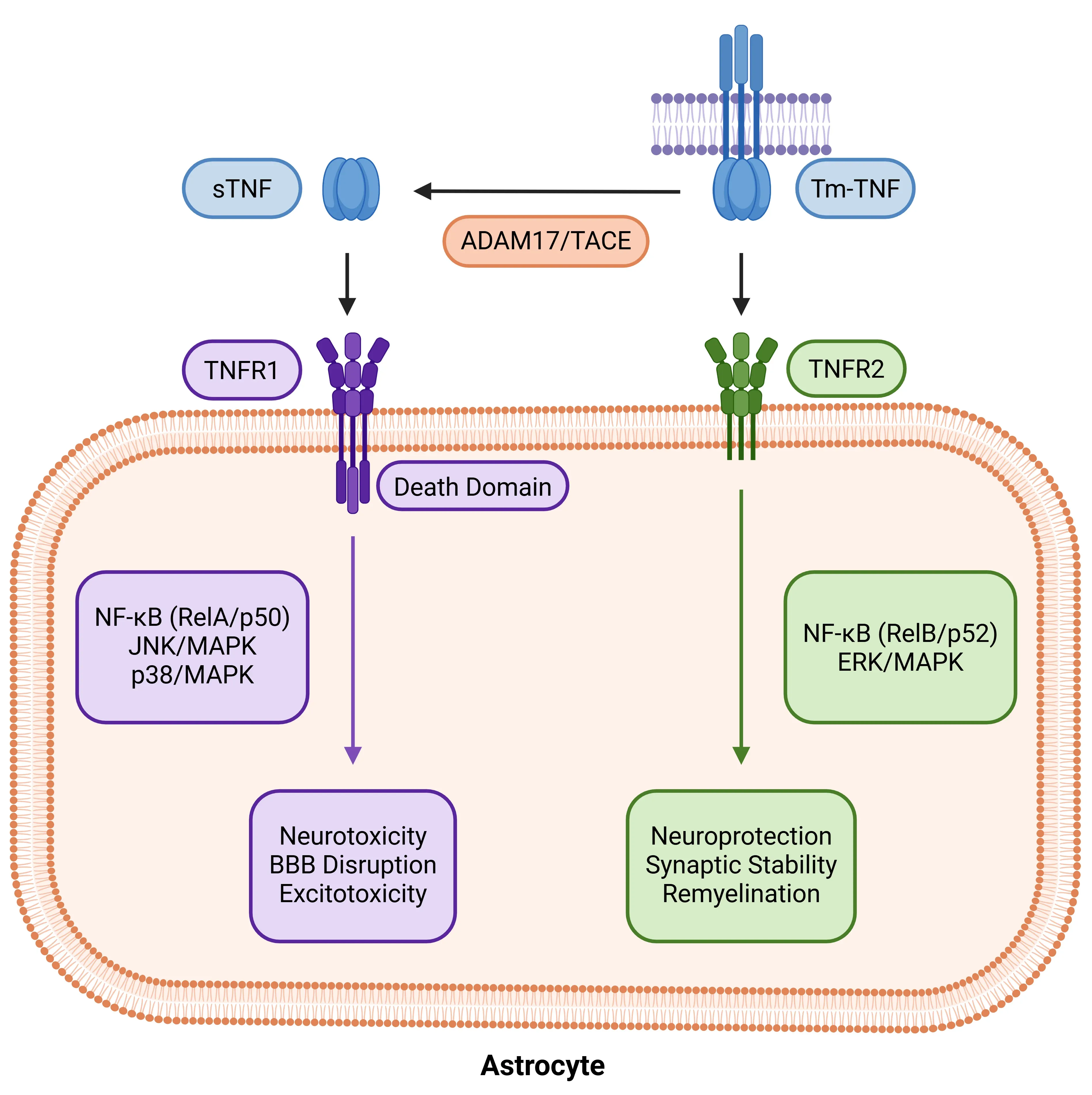
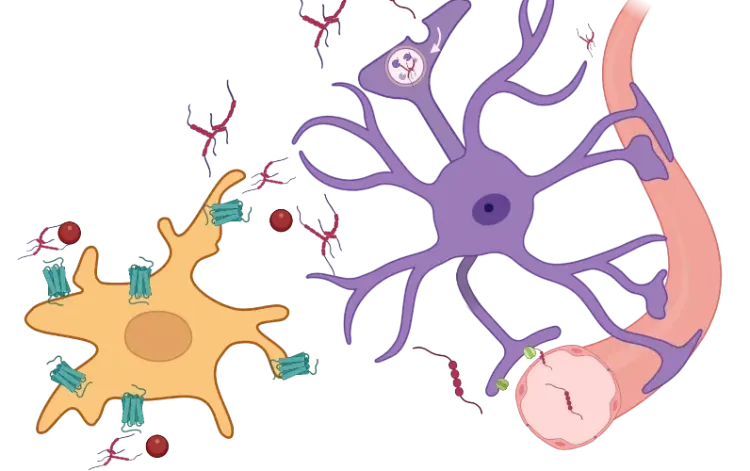
TNF-α receptor dichotomy in astrocytes. Astrocytic responses to TNF-α depend on ligand form and receptor specificity. sTNF cleaved from tmTNF by ADAM17/TACE activates TNFR1, triggering NF-κB (RelA/p50), JNK, and p38 MAPK pathways that drive neurotoxicity and BBB disruption. In contrast, tmTNF preferentially binds TNFR2, signaling via non-canonical NF-κB (RelB/p52) and ERK/MAPK to promote neuroprotection, synaptic stability, and remyelination. This dichotomy positions TNF-α as a key regulator of astrocytic state between injury and repair.
What roles do reactive astrocytes play in TNF-α–mediated neuroinflammation?
Reactive astrocytes under TNF-α adopt multiple harmful roles that extend beyond simple glial activation. Here, we describe how these states contribute to neuroinflammation, vascular dysfunction, and direct neuronal injury across different disease contexts.
Glial Toxicity and Vascular Dysfunction
Astrocytic reactivity is profoundly shaped by TNF-α, which functions as a key amplifier of microglia–astrocyte communication in the inflamed brain. Microglia-derived TNF-α, together with IL-1α and C1q, is sufficient to convert homeostatic astrocytes into neurotoxic A1 states both in vitro and in vivo, establishing TNF-α as a key inducer of astrogliosis. Neutralization of this IL-1α/TNF/C1q triad prevents A1 conversion and preserves neuronal and oligodendrocyte survival, confirming that TNF-α-dependent reactivity is a cause of pathology rather than just a byproduct of disease (Liddelow, 2017). In hippocampal circuits, TNF-α activation of astrocytic TNFR1 initiates an astrocyte-neuron cascade that remodels excitatory synapses and impairs contextual memory (Habbas, 2015).
At the vascular interface, TNF drives astrocytes into a STAT3-dependent inflammatory reactive state, causing them to release factors that disrupt BBB integrity and induce vascular endothelial inflammation (Kim, 2022). Building on this evidence of vascular disruption, recent human brain-chip studies demonstrate that TNF-primed astrocytes not only destabilize barrier function but also impair microvascular morphogenesis, a defect reversible with MEK1/2 inhibition (de Rus Jacquet, 2023).
Taken together, TNF-α-reactive astrocytes act as key drivers of dysfunction by fueling toxic glial states, disrupting synaptic communication, and weakening BBB and microvascular integrity, thereby amplifying neuroinflammatory damage.
Inflammatory and Oxidative Drivers of Neuroinflammation
Once astrocytes adopt a TNF-α-driven reactive state, they become active sources of inflammation and oxidative stress that spread damage well beyond the glial compartment. A central mechanism is activation of the NF-κB pathway, which induces production of pro-inflammatory mediators:
These signals recruit leukocytes and amplify local CNS immune activity (Giovannoni, 2020).
In parallel, reactive astrocytes produce reactive oxygen and nitrogen species (ROS/NOS), linking TNF-α signaling to oxidative stress pathways that further destabilize neuronal and vascular homeostasis (Ding, 2021). Bidirectional crosstalk with microglia intensifies these responses: astrocytic NF-κB activation is sufficient to drive microglial proliferation and leukocyte infiltration, showing that astrocytes actively escalate neuroinflammation rather than passively respond to it (Ouali Alami, 2018).
See: Mitochondrial Dysfunction in Microglia & Astrocytes
Human iPSC-derived astrocyte models replicate these features, as combined IL-1β and TNF-α stimulation induces NF-κB nuclear translocation, morphological remodeling, and acquisition of an immune-reactive phenotype (Hyvärinen, 2019). Consistent with these experimental models, single-nucleus RNA-seq (snRNA-seq) from human AD and MS tissues demonstrates that NF-κB-driven astrocyte states cluster around plaques and lesions, highlighting their direct clinical relevance (Escartin, 2021).
Together, these findings show that TNF-α-reactive astrocytes function as potent engines of inflammation and oxidative stress, actively amplifying neuroinflammatory cascades and worsening neuronal and vascular vulnerability.
Neurotoxicity and Circuit Dysfunction
TNF-α–driven astrocyte reactivity not only strips away their normal supportive roles but also makes them active sources of neuronal and oligodendroglial injury. Functionally, A1 astrocytes lose their normal pro-synaptogenic support and secrete factors that kill neurons and oligodendrocytes, directly linking TNF-α-induced reactive states to neurodegeneration (Liddelow, 2017). One mechanism involves the secretion of saturated lipids in APOE/APOJ lipoparticles; blocking the lipid-metabolizing enzyme ELOVL1, prevents this toxicity, connecting astrocyte lipid metabolism to TNF-induced neuronal death (Guttenplan, 2021).
At the circuit level, sustained TNF-α exposure, alone or combined with IL-6, disrupts neuronal firing patterns in human neuron–astrocyte co-cultures, leading to abnormal rhythms that may underlie cognitive and behavioral deficits during inflammation (Goshi, 2025). Similar disruptions have been observed in vivo, where chronic TNF-α exposure drives hippocampal hyperexcitability and epileptiform activity (Vezzani, 2020).
Overall, TNF-α–reactive astrocytes emerge as critical drivers of neuronal loss and network instability, directly connecting glial reactivity to the progression of neurodegenerative disease.
Together, the evidence demonstrates that TNF-α fundamentally reshapes astrocyte biology, shifting them from guardians of CNS homeostasis into central engines of dysfunction. By positioning astrocytes at the crossroads of inflammation, vascular compromise, and neuronal vulnerability, TNF-α reveals them not as passive responders but as decisive regulators of neurodegenerative progression. This perspective reframes astrocytes from background players to pivotal therapeutic targets in the fight against chronic neuroinflammation.
How does TNF-α contribute to neurodegenerative disease progression through astrocytes?
Chronic TNF-α exposure gradually pushes astrocytes into harmful states that alter their gene expression, structure, and metabolism. Even at low levels, persistent TNF-α signaling causes astrocyte enlargement, inflammatory gene rewiring, and metabolic stress, leading researchers to view astrocyte reactivity not as simply “protective or toxic,” but as a spectrum of shifting states (Escartin, 2021).
Inflammatory microglial cues containing TNF-α reinforce a transition toward neurotoxic A1-like astrocytes, providing a direct link between chronic innate immune activation and astrocyte dysfunction in neurodegenerative disease (Liddelow, 2017). Human iPSC-derived astrocytes replicate this process, where TNF-α (±IL-1β) rapidly activates NF-κB, and remodels morphology and inflammatory gene networks (Hyvärinen, 2019). In PD-relevant human astrocytes, TNF-α or α-synuclein fibrils drive astrocytes into overlapping immune-metabolic states marked by defective mitochondrial respiration, showing how prolonged cytokine exposure undermines cellular energy balance (Russ, 2021).
Together, these findings show that chronic TNF-α signaling imprints a persistent inflammatory and metabolic burden on astrocytes, shifting them into dysfunctional states that weaken their supportive roles and actively contribute to neurodegenerative disease progression.
At the synaptic level, TNF-α alters astrocytic glutamate handling and gliotransmission in ways that destabilize neuronal circuits and heighten excitotoxic vulnerability:
- Activation of astrocytic TNFR1 alone can alter excitatory transmission and impair hippocampal memory in autoimmune demyelination, demonstrating a direct causal role for astrocytic TNF-α in network dysfunction (Habbas, 2015).
- TNF-α, via the TNFR1-NF-κB pathway, reduces the expression of EAAT2/GLT-1 transporters, weakening glutamate uptake and exposing motor neurons to toxic overstimulation (Jiang, 2019).
- Human iPSC-derived astrocytes co-stimulated with TNF-α and IL-1β show impaired glutamate clearance and stronger inflammatory activation (Hyvärinen, 2019).
- TNF-α enhances gliotransmission in cerebellar circuits by increasing Bergmann glia glutamate release, which raises Purkinje cell excitability through mGluR1 signaling, destabilizing firing patterns (Shim, 2018).
- Sustained TNF-α (±IL-6) exposure disrupts neuronal firing dynamics in human neuron-astrocyte co-cultures on microelectrode arrays (MEAs), indicating chronic cytokines drive emergent circuit dysrhythmias (Goshi, 2025).
- In vivo studies show that TNF-α overexpression increases seizure susceptibility and accelerates cognitive decline, linking astrocytic dysfunction to broader network instability (Vezzani, 2020).
Together, these findings demonstrate that TNF-α–driven astrocytic dysfunction undermines glutamate regulation and synaptic balance, leading to widespread circuit instability and progressive cognitive impairment.
Within the neurovascular unit (NVU), TNF-α-primed astrocytes transmit inflammatory cues that weaken BBB integrity and impair vascular function. In human and ex vivo models, TNF-α drives astrocytes into a STAT3-dependent reactive state marked by upregulation of SERPINA3 (also known as α1-antichymotrypsin, an acute-phase serine protease inhibitor), which reduce BBB integrity and promotes endothelial dysfunction (Kim, 2022).
Astrocytes carrying the PD-linked LRRK2 mutation adopt pro-inflammatory profiles and fail to support microvascular morphogenesis in a human brain-chip BBB system; this deficit is reversed by MEK1/2 inhibition (de Rus Jacquet, 2023). These findings align with broader evidence that astrocytic endfeet and their secreted molecules regulate BBB stability, and that inflammatory remodeling of these interfaces contributes to vascular dysfunction in neurodegeneration (Yue, 2023). Consistent with these mechanistic insights, spatial transcriptomics in human MS lesions demonstrates that TNF-primed astrocytes cluster at perivascular regions, producing cytokine-rich niches that actively destabilize BBB structure (Mazziotti, 2024).
These findings demonstrate that TNF-α–driven astrocytes actively disrupt BBB integrity and vascular health, making them key contributors to neurovascular dysfunction in neurodegenerative disease.
Across major neurodegenerative disorders, TNF-α-driven astrocytic programs act as common amplifiers of disease progression. By coupling inflammatory signaling to synaptic, metabolic, and vascular dysfunction, astrocytes transform TNF-α cues into local mechanisms that accelerate neuronal vulnerability and tissue decline. While these core processes are shared across disorders, the ways in which TNF-α-reactive astrocytes interact with disease-specific environments vary considerably. In the following sections, we describe how TNF-α signaling through astrocytes contributes to progression in Alzheimer’s disease, Parkinson’s disease, ALS, and multiple sclerosis (MS).
Alzheimer’s Disease (AD)
TNF-α has emerged as a pivotal mediator in AD, linking amyloid-β pathology, neuroinflammation, and synaptic dysfunction. Elevated levels of TNF-α have been consistently reported in both cerebrospinal fluid (CSF) and serum of patients with mild cognitive impairment (MCI) and AD, where higher baseline concentrations predict faster cognitive decline and increased risk of progression to dementia (Kim, 2017; Lista, 2024). Importantly, these biochemical changes are paralleled by astrocytic alterations. Reactive astrocyte biomarkers, including CSF GFAP and YKL-40, mediate the effects of amyloid-β and tau on hippocampal atrophy and cognition, linking astrocyte reactivity to downstream neurodegeneration in living humans (Ferrari-Souza, 2022; Pelkmans, 2024). Neuropathological studies further reveal that reactive astrocytes cluster around Aβ plaques and neurofibrillary tangles, penetrating plaque cores and contributing both to containment and propagation of pathology (Perez-Nievas, 2018).
For an analysis of astrocyte morphology in the amyloid-β plaque microenvironment, see: Astrocytes & Amyloid-β Mouse Models of Alzheimer's Disease
A central mechanism by which TNF-α contributes to AD involves its ability to drive amyloidogenic processing in astrocytes. Specifically, TNF-α signaling:
- Upregulates amyloid precursor protein (APP) and BACE1 expression, accelerating Aβ generation and plaque accumulation (Zhao, 2011).
- Disrupts synaptic homeostasis by shifting the excitatory–inhibitory balance:
- Enhances glutamatergic transmission
- Reduces GABAergic inhibition
- Increases AMPA receptor surface trafficking,
These effects collectively predispose hippocampal circuits to excitotoxicity (Pribiag, 2013; Heir, 2020). This excitotoxic injury is further amplified by astrocyte-mediated glutamate release, which compromises neuronal survival (Santello, 2011).
Astrocytes themselves are not passive responders but key drivers of TNF-α signaling in AD. Experimental evidence shows:
- TNF-α signaling persists in hippocampal cultures lacking microglia, but is abolished with astrocyte-specific TNF deletion, establishing astrocytes as indispensable for activity-dependent TNF release (Heir, 2024).
- Astrocytic NF-κB activation regulates TNF production in response to glutamate spillover, positioning astrocytes at the center of cytokine-driven circuit remodeling.
- Modulation of astrocyte NF-κB signaling alters neuronal and synaptic outcomes in AD models (Jong Huat, 2024).
- Impairments in astrocytic glutamate handling, particularly reduced EAAT2/GLT-1 transporter function, converge with TNF-driven excitotoxicity to exacerbate neurodegeneration (Wood, 2022).
Genetic studies reinforce the pathogenic role of TNF-α. The G-308A polymorphism in the TNF-α promoter increases transcriptional activity and protein expression, with several meta-analyses linking it to heightened AD susceptibility (Wang, 2015). This variant may act synergistically with the APOE-ε4 genotype to accelerate inflammatory priming and disease progression (Contreras, 2020), underscoring the importance of TNF signaling as a common denominator across sporadic and familial AD.
Therapeutic studies highlight the translational potential of targeting TNF-α. Data show:
- Patients with systemic inflammatory diseases treated with TNF blockers, such as Etanercept or Adalimumab, have reduced dementia incidence compared with untreated populations.
- Small pilot trials and case reports using peri-spinal Etanercept delivery report rapid and sometimes sustained cognitive improvements in AD patients.
- Larger randomized controlled trials with subcutaneous Etanercept failed to demonstrate significant benefits, likely due to poor BBB penetration of large biologics.
- Preclinical studies with BBB-penetrant TNF inhibitors show reduced Aβ and tau pathology alongside cognitive improvements in AD mouse models, strengthening the rationale for TNF-targeted therapy (Plantone, 2024).
Taken together, these findings demonstrate that TNF-α, through astrocyte-driven mechanisms, is a central driver of amyloid-β accumulation, excitotoxicity, and neurodegeneration in AD, making it a biologically plausible therapeutic target. While clinical data remain preliminary, particularly regarding BBB delivery, TNF-α inhibition represents a promising avenue for disease modification.
Parkinson’s Disease (PD)
Astrocytic TNF-α plays a central role in Parkinson’s disease (PD), emerging as a mediator that connects α-synuclein pathology with neuroinflammatory amplification and neuronal vulnerability. In patients with PD, TNF-α and soluble receptor are consistently elevated in brain, CSF, and blood, correlating with dopaminergic neurodegeneration, cognitive decline, and overall disease severity (Liu, 2022). Accumulation of α-synuclein within astrocytes not only disrupts mitochondrial and endoplasmic reticulum function, but also drives a reactive, pro-inflammatory phenotype (Wang, 2021). Importantly, astrocytes containing α-synuclein inclusions secrete high levels of TNF-α, positioning this cytokine as a central effector linking proteinopathy to inflammatory amplification (Lee, 2010).
Astrocyte-derived TNF-α contributes to a feed-forward loop of glial reactivity in PD.
For an in-depth review of how α-synuclein influences microglia and astrocytes in PD and other synucleinopathies, see: Microglia, Astrocytes & α-Synuclein in Parkinson’s Disease
Alongside IL-1β and IL-6, astrocytic TNF-α:
- Amplifies microglial activation, thereby accelerating dopaminergic neuronal loss.
- Exacerbates neuronal vulnerability.
- Establishes a feed-forward loop of glial reactivity linking astrocyte-driven cytokine signaling to progressive nigrostriatal degeneration (Wang, 2023).
Experimental PD models show that both IFN-γ and TNF-α are required to sustain microglial and astroglial activation even before overt neuronal loss, underscoring TNF-α’s role in initiating glial-driven neuroinflammation (Barcia, 2011). Mechanistic studies further reveal that the regulator RGS5:
- Shifts TNFR2 signaling from protective to pro-inflammatory in astrocytes.
- Amplifies TNFR1 activation.
- Enhances α-synuclein aggregation, neurodegeneration, and mortality in PD models (Yin, 2023).
Conversion of astrocytes into a neurotoxic A1 state represents another pathological mechanism in which TNF-α is indispensable. Microglia release IL-1α, TNF-α, and C1q, which together convert astrocytes into this harmful phenotype. A1 astrocytes have been identified in the postmortem brains of PD patients (Liddelow, 2017). In PD animal models (α-syn PFF model of sporadic PD and in the hA53T Tg mouse model), pathological α-synuclein also promotes A1 astrocyte formation, and blocking this process protects dopaminergic neurons and preserves motor function (Yun, 2018). These A1 astrocytes:
- Lose the ability to provide trophic and synaptic support.
- Secrete neurotoxic factors, including complement components and ROS.
- Exacerbate neuronal injury (Liddelow, 2017).
Human studies confirm the pathogenic role of astrocytic TNF-α in PD. Single-nucleus RNA-seq of human PD midbrain reveals pan-glial activation with a CD44high astrocyte phenotype and cytokine-signaling trajectories, consistent with chronic inflammatory tone in the nigral niche (Smajić, 2022). In cultured human astrocytes, TNF-α and α-synuclein fibrils trigger immune-reactive states with impaired mitochondrial respiration, highlighting TNF-responsive metabolic liabilities that align with PD pathogenesis (Russ, 2021). This pathogenic cascade is further amplified by complement C4, which:
- Boosts astrocytic pro-inflammatory responses to α-synuclein.
- Promotes neuronal apoptosis and synaptic pathology (Zou, 2025).
Additionally, inflammation-primed human astrocytes impair microvascular morphogenesis and disrupt BBB integrity in brain-chip models (de Rus Jacquet, 2023). Mechanistically, TNF drives astrocytes into a STAT3-dependent acute-phase-like state that conveys systemic inflammation to the brain endothelium, thereby promoting BBB breakdown in PD (Kim, 2022).
Taken together, these findings position astrocytic TNF-α as a central mechanism that connects α-synuclein pathology, glial amplification, synaptic dysfunction, and vascular impairment in PD.
Amyotrophic Lateral Sclerosis (ALS)
Astrocytic TNF-α signaling has emerged as an early and persistent driver of motor neuron vulnerability in ALS. Elevated levels of TNF-α are consistently detected in the CSF and blood of ALS patients, correlating with disease progression (Jiang, 2022). Similarly, in SOD1G93A mice, TNF-α and its receptors are upregulated in the spinal cord of even before symptom onset, suggesting that this pathway contributes to early pathogenesis (Brambilla, 2016). In astrocyte-motor neuron co-cultures derived from these models, membrane-bound TNF-α increases in motor neurons while TNFR2 levels decrease, creating an imbalance in receptor signaling. Notably:
- Deleting TNFR2, but not TNFR1, rescues motor neuron survival, identifying the mTNF-α–TNFR2 axis as a key mediator of neurotoxicity (Tortarolo, 2015).
- Conversely, TNFR1 signaling may exert a protective effect, possibly through astrocytic release of glial-derived neurotrophic factor (GDNF), since its ablation worsens outcomes (Brambilla, 2016).
These findings underscore that the contributions of TNFR1 and TNFR2 to ALS pathology are highly context-dependent, varying between isolated co-culture systems and complex in vivo environments. Further, both TNFR1 and TNFR2 activate the apoptosis signal-regulated kinase 1 (ASK1)/p38MAPK pathway, which is critically involved in motor neuron death. Inhibiting p38 MAPK protects motor neurons in SOD1G93A mouse-derived astrocyte/MN co-culture models, highlighting this pathway as a key downstream effector of TNF signaling in ALS (Dewil, 2007). In parallel:
- In astrocytes carrying mutant FUS, TNF-α activates NF-κB and alters AMPA receptor subunit composition by reducing GluA2 incorporation.
- This change increases calcium permeability and excitotoxic stress in motor neurons (Kia, 2018).
- Genetic TNF-α loss or pharmacological neutralization in FUS-ALS models rescues motor behavior and neuron survival, identifying astrocyte-derived TNF-α as the proximate toxic signal (Jensen, 2022).
Astrocytic TNF-α also shapes the inflammatory milieu, driving both glial and immune responses that worsen ALS pathology. Key mechanisms include:
- Inducible NF-κB activation in astrocytes promotes microglial proliferation and peripheral leukocyte recruitment, hastening symptomatic progression (Ouali Alami, 2018).
- The microglial IL-1α/TNF/C1q signaling triad induces a neurotoxic A1 astrocyte phenotype, and disrupting this axis prolongs survival in SOD1G93A mice (Guttenplan, 2020).
- Astrocytes carrying SOD1 mutations adopt a reactive, dysfunctional state driven by mitochondrial stress, proteasome impairment, and innate immune activation, which TNF-α signaling exacerbates.
- Although fibroblast growth factor 4 (FGF4) can transiently restore astrocytic homeostasis, TNF-α overrides this protection by sustaining NF-κB activity, leaving motor neurons vulnerable (Velasquez, 2024).
Astrocytic TNF-α disrupts glutamate homeostasis, further enhancing excitotoxic stress in ALS. Specifically, TNF-α signaling via astrocytic TNFR1:
- Suppresses EAAT2/GLT-1 expression.
- Drives glutamate release.
- Leads to impaired glutamate clearance and excitotoxic stress on motor neurons, a mechanism consistent with human ALS pathology (Jiang, 2019).
This disruption synergizes with TNF-induced changes in AMPA receptor composition, reinforcing calcium-driven excitotoxicity (Kia, 2018; Jensen, 2022).
Taken together, astrocytic TNF-α acts as a convergence point linking inflammation, receptor imbalance, and excitotoxicity in ALS. By simultaneously amplifying inflammatory cascades, altering glutamate handling, and increasing excitatory receptor permeability, TNF-α accelerates motor neuron degeneration and drives disease progression.
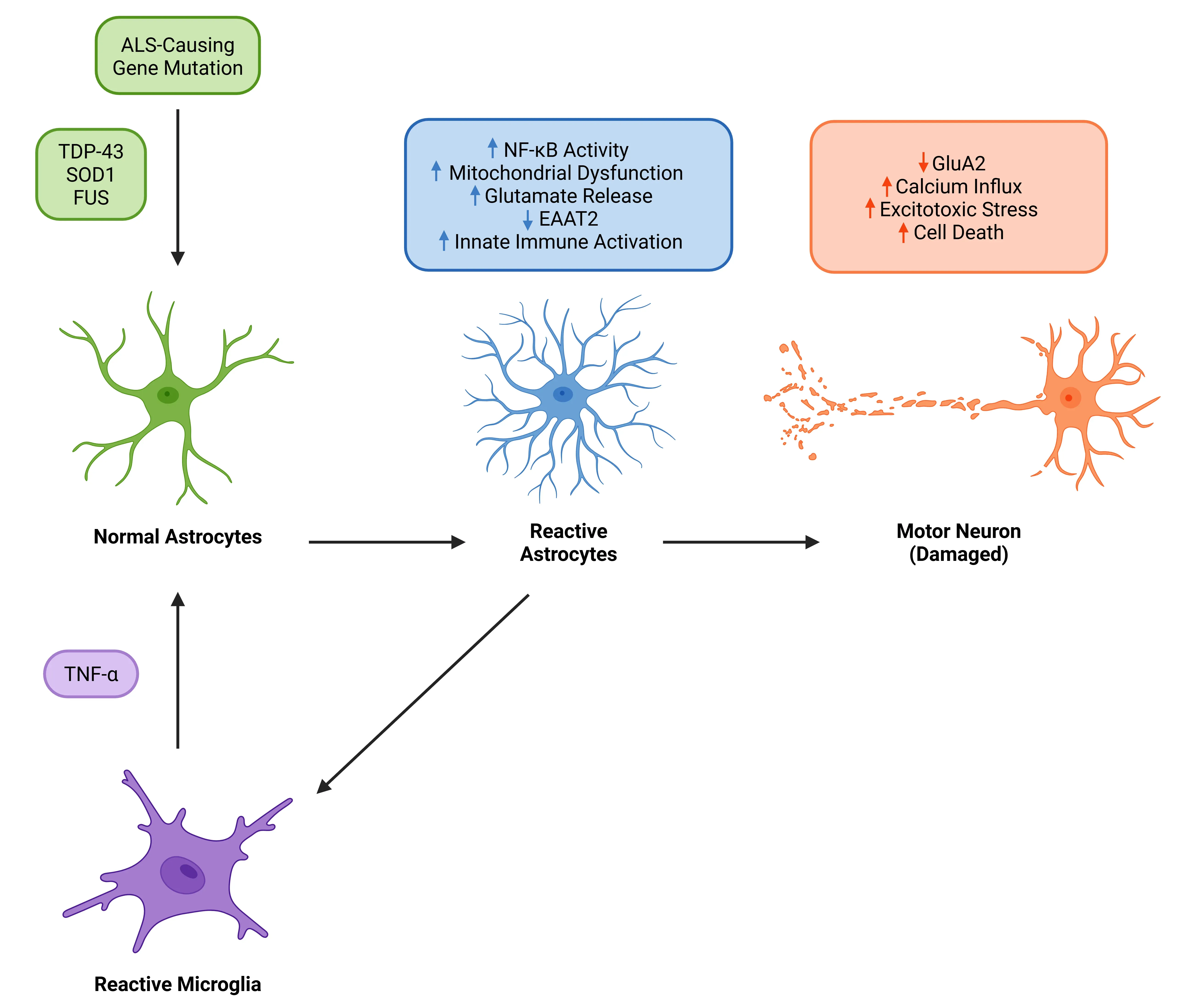
TNF-α as a pivotal driver of astrocytic reactivity and motor neuron degeneration in ALS. In ALS associated with mutations in TDP-43, SOD, or FUS, TNF-α released by reactive microglia promotes astrocyte reactivity via TNFR signaling. This activates NF-κB in astrocytes and impairs glutamate clearance, leading to excitotoxic stress and motor neuron cell death. Reactive astrocytes further amplify neuroinflammation, creating a feedback loop that accelerates motor neuron loss.
Multiple Sclerosis (MS)
In multiple sclerosis (MS), astrocytes emerge as critical mediators of the effects of TNF-α, linking inflammatory cascades with both neurotoxic and reparative processes. TNF-α is consistently elevated in the cerebrospinal fluid (CSF) and detected in both acute and chronic active MS lesions, where higher levels correlate with worse disability and accelerated disease progression (Sharief, 1991; Kosa, 2022). Lesion-level pathology confirms TNF upregulation in acute and chronic active plaques, aligning TNF signaling with sites of active tissue damage (Mazziotti, 2024).
A key mechanism by which astrocytes amplify local TNF signaling involves the proteolytic processing of membrane-bound TNF. Within active MS lesions, reactive astrocytes upregulate ADAM17/TACE, which cleaves tmTNF into its soluble form. This shift preferentially activates TNFR1, enhancing apoptotic and pro-inflammatory cascades (Plumb, 2006). In experimental autoimmune encephalomyelitis (EAE), astrocytic TNFR1 signaling was shown to drive hippocampal synaptic remodeling and cognitive impairment, directly linking glial TNF signaling to network dysfunction (Habbas, 2015). Follow-up genetic rescue studies reinforced that reinstating TNFR1 specifically in astrocytes is sufficient to re-establish the synaptic and behavioral EAE phenotype (Di Castro, 2022). Together, these findings position astrocytes not merely as responders but as amplifiers of TNF-mediated neurotoxicity.
At the same time, astrocytes are also capable of harnessing protective TNF pathways, underscoring their dual role in MS. They express both TNFR1 and TNFR2, and mounting evidence shows that TNFR2 signaling favors neuroprotection and repair. In demyelinating models, astrocytic TNFR2 activation promotes anti-inflammatory phenotypes and supports remyelination through mechanisms such as pCXCL12-driven oligodendrocyte recruitment (Brambilla, 2011; Patel, 2012). Recent studies add that TNFR2 constrains pro-inflammatory astrocytic programs and enhances trophic support for myelination, highlighting it as a counterbalance to TNFR1-mediated damage (Raphael, 2019; Pegoretti, 2023).
These mechanistic insights help explain why clinical attempts at broad anti-TNF therapy in MS have failed:
- Non-selective TNF blockade has been associated with:
- Paradoxical exacerbation of demyelinating events
- De novo CNS inflammatory syndromes (W. Xie, 2024)
Instead, the therapeutic direction has shifted toward selective modulation of TNF signaling (Brambilla, 2011; Pegoretti, 2023):
- Promising strategies in both preclinical and translational studies include:
- Inhibiting TNFR1
- Neutralizing sTNF
- Sparing or agonizing TNFR2
Taken together, these findings position TNF as a pivotal cytokine orchestrating astrocytic responses in MS, constantly tipping the balance between injury and repair. The dual nature of astrocytic TNF signaling highlights the challenge and opportunity of therapeutic targeting, making receptor-selective modulation an attractive strategy for neuroprotection and remyelination.
What therapeutic strategies target TNF-α signaling in astrocytes?
Current therapeutic strategies can be grouped into four major approaches, each targeting a distinct aspect of TNF-α signaling in astrocytes:
TNF-α-Receptors/Ligands Inhibitors
Therapeutic strategies increasingly aim to fine-tune TNF-α signaling in astrocytes by suppressing pathological TNFR1 activity while preserving or enhancing the reparative functions of TNFR2. This balance is critical because selective inhibition of TNFR1 or neutralization of sTNF can spare tmTNF-TNFR2 signaling, which supports trophic and remyelinating programs within the CNS (Fischer, 2020).
Key approaches include:
- Receptor-Selective Antagonists
- Atrosimab, a monovalent human TNFR1-specific antagonist, has demonstrated efficacy in inflammatory models including the EAE model, validating selective TNFR1 blockade as a relevant astrocyte-centered intervention.
- Additional preclinical studies extend these benefits to acute neurodegeneration, underscoring the broader neuroprotective potential of this drug class (Ortí-Casañ, 2023).
- Ligand-Targeted Strategies:
- Dominant-negative TNF and XPro1595 (pegipanermin) neutralize sTNF while preserving mTNF-TNFR2 signaling (MacPherson, 2017; De Sousa Rodrigues, 2019).
- ADAM17/TACE inhibition prevents cleavage of tmTNF into sTNF, further favoring TNFR2 pathways (L. Xie, 2024).
Despite these promising results, translation to the clinic remains challenging because most biologics have poor penetration across the BBB. This limitation has fueled the development of molecular shuttles and CNS-targeted delivery systems (Kouhi, 2021). In addition, clinical experience with non-selective systemic TNF inhibitors has shown paradoxical worsening of demyelinating conditions, underscoring the importance of receptor- and ligand-selective strategies in neuroinflammatory settings (Mazziotti, 2024).
Targeting Downstream Signaling
Another approach involves modulating signaling pathways downstream of TNF receptors, particularly NF-κB and MAPK, which regulate astrocyte reactivity.
- Key Downstream Targets:
- NF-κB: Astrocyte-specific activation amplifies neuroinflammation in vivo, suggesting that selective inhibition may be beneficial, though new human-cell studies show its effects on neurons can be both protective and harmful depending on the context (Giovannoni, 2020).
- p38α MAPK: Selective inhibitors such as MW150 suppress neuroinflammation and improve cognition in AD models, suggesting translational potential (Frazier, 2024).
- MEK1/2 (ERK pathway): Inhibition restores microvascular morphogenesis in astrocytes with PD-associated mutation. Under normal conditions, the MEK/ERK pathway helps astrocytes regulate stress responses and mount balanced inflammatory signaling. However, in disease this protective role becomes pathologically amplified, highlighting MEK/ERK as a tractable lever for BBB stabilization and a promising therapeutic target in neurodegeneration (de Rus Jacquet, 2023).
- JNK: Inhibition limits astrocytic CXCL1 release and downstream neuronal stress, positioning JNK as a parallel pathway target (Zhang, 2022).
Modulating Astrocyte Phenotypes
A complementary therapeutic strategy aims to reprogram astrocytes away from neurotoxic phenotypes and toward homeostatic or reparative states. Blocking the IL-1α/TNF/C1q axis prevents conversion to A1-like astrocytes and preserves neuronal and oligodendrocyte viability in vivo, providing one of the most compelling demonstrations that astrocyte phenotype modulation is therapeutic (Liddelow, 2017). Other promising mechanisms include:
- ELOVL1 inhibition, which blocks release of toxic lipid particles, highlighting lipid metabolism as another druggable vulnerability (Guttenplan, 2021).
- HDAC3 inhibitors, which reduce inflammatory gene activation while sparing supportive astrocyte functions (Clayton, 2024).
- GLP-1 receptor agonists (g. NLY01), which suppress microglial-induced astrocyte toxicity and are in ongoing clinical trials in AD and PD (Yun, 2018; Park, 2021).
Gene Therapy and Novel Biologics
Gene and biologic therapies further are expanding options for astrocyte-specific control of TNF signaling:
- IL-2 gene delivery to astrocytes increases local regulatory T cells and reduces neuroinflammation without affecting systemic immunity (Yshii, 2022).
- Advances in AAV vectors using GFAP promoters, next-generation astrocyte-specific elements, and broad serotype enhancers, now make astrocyte-selective gene transfer feasible in both rodents and larger species (O’Carroll, 2021; Heffernan, 2022; Gleichman, 2023).
- Combination strategies, such as sequential TNFR2 agonism followed by TNFR1 antagonism, improves outcomes in humanized EAE models and demonstrate that temporal biasing of receptor activity can maximize therapeutic efficacy while minimizing risk (Pegoretti, 2023).
Overall, targeting astrocytic TNF-α signaling highlights the need to suppress its pathological inflammatory outputs while preserving protective functions that support repair and BBB stability. The key barrier remains limited brain penetration of current therapies, but advances in selective inhibitors, delivery systems, and astrocyte-focused interventions raise the prospect of turning TNF-α modulation into a viable neuroprotective treatment.
How does TNF-α compare to other cytokines in neurodegeneration?
Several key distinctions set TNF-α apart from other cytokines in neurodegeneration, shaping both its role in disease progression and its potential as a therapeutic target.
Distinctive Functions of TNF-α (Apoptosis, Necroptosis, Inflammation)
TNF-α occupies a unique position among neuroinflammatory cytokines because its receptor wiring links inflammatory signaling to programmed cell death. Unlike other mediators, TNF-α couples gene induction to apoptosis and necroptosis through TNFR1, engaging RIPK1/RIPK3/MLKL pathways in addition to NF-κB-driven survival and pro-inflammatory outputs (Holbrook, 2019; van Loo, 2023).
By contrast:
- IL-6 signals through:
- “Classic” signaling via membrane IL-6R, which promotes protective and regenerative responses.
- “Trans-signaling” via soluble IL-6R, which amplifies inflammation.
- Soluble IL-6 receptors act mainly as agonists, contrasting with TNF, where soluble receptors tend to be antagonistic (Rose-John, 2021).
- Activates STAT3-biased responses, amplifies astrocyte reactivity, and disrupts vascular stability, contributing to BBB breakdown (Mora, 2024).
- Interferon-γ (IFN-γ) reprograms astrocytes toward antigen presentation, producing interferon-responsive reactive astrocytes (IRRAs) instead of TNF-induced pro-death states (Rostami, 2020; Prakash, 2024; Lee, 2023).
These findings suggest that TNF-α uniquely bridges innate inflammatory cues with executioner death pathways, positioning it closer to “final common” routes of neurodegeneration than IL-6 or IFN-γ.
TNF-α Synergy and Divergence with Other Cytokines in Astrocytes
Functionally, TNF-α often acts as a priming cytokine that prepares astrocytes for maladaptive phenotypes, especially when acting together with other mediators.
Key interactions include:
- TNF-α + IL-1α + C1q: Drives astrocytes into A1-like states, leading to synapse-supporting function loss and death of neurons and oligodendrocytes (Liddelow, 2017).
- TNF-α + IL-1β: Strong activation of NF-κB signaling in human iPSC-derived astrocytes, reshaping their structure and inducing a powerful immune-reactive state (Hyvärinen, 2019).
These findings highlight that while TNF-α synergizes with IL-1α (together with C1q) and with IL-1β to drive neurotoxic and immune-reactive astrocyte states, cytokines such as IFN-γ and IL-6 activate divergent pathways, emphasizing how distinct cytokine programs shape astrocyte behavior in disease.
Spatial and Single-Cell Signatures of TNF-Driven Astrocytes
Single-cell and spatial transcriptomics show that TNF-α-responsive astrocytes form unique, disease-relevant states:
- In AD:
- snRNA-seq shows astrocytes located next to plaques lose their normal supportive gene programs and instead switch on complement and inflammatory pathways driven by TNF (Dai, 2023).
- Spatial transcriptomics further confirm that these plaque-border astrocytes are strongly shaped by TNF/IL-1 signaling and cluster around degenerating neurons (He, 2024).
- Meta-analyses show parallel increases of TNF-α, IL-6, and IL-1β along the AD continuum, which directly correlate with worsening cognitive decline (Lista, 2024; Serna, 2025).
- In PD:
- snRNA-seq demonstrates widespread activation of glial cells, including a subset of CD44-high astrocytes whose gene expression reflects chronic TNF/IL-1-driven inflammation in the substantia nigra (Smajić, 2022).
Therapeutic Selectivity of TNF-α Versus Other Cytokines
TNF-α stands out therapeutically because its signaling architecture allows selective targeting of harmful TNFR1 pathways while sparing protective TNFR2 functions. This kind of selective balance is unique to TNF and cannot be achieved with IL-6 or IFN-γ blockade (Fischer, 2020; Papazian, 2021).
- Non-selective anti-TNF therapies can worsen CNS autoimmunity (Mazziotti, 2024).
- IL-6 therapies (g. sgp130Fc) block inflammation while leaving protective “classic” IL-6 signaling intact, but do not address the cell-death pathways unique to TNF receptor signaling (Rose-John, 2021).
- IFN-γ- therapies modulate immune-related astrocyte functions such as antigen presentation, rather than the apoptotic and necroptotic cascades driven by TNF (Prakash, 2024; Lee, 2023).
Together, these comparisons show that TNF-α is unique among cytokines because its signaling can be therapeutically fine-tuned: selective blockade of TNFR1 or sTNF suppresses neurotoxic and cell-death pathways while preserving TNFR2-mediated repair, a therapeutic balance that IL-6 or IFN-γ interventions cannot achieve.
Overall, evidence shows that TNF-α shapes astrocyte behavior in ways that are more directly linked to neurodegenerative progression than other cytokines. By coupling inflammatory cues to cell-death pathways, amplifying maladaptive astrocyte states, and driving region-specific transcriptional changes, TNF-α places astrocytes at the center of both disease mechanisms and therapeutic opportunity.
Our team would be happy to answer any questions about the function of TNF-α in astrocytes and its contribution to neurodegenerative diseases or provide specific information about the AD, ALS, and PD models we use for therapeutic efficacy studies.
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on TNF-α and microglia in neurodegenerative diseases and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
TNF-α (TNF-alpha) & Microglia in Neurodegenerative Diseases
An overview of the function of tumor necrosis factor-alpha (TNF-α) in microglia and its contribution to the progression of neurodegeneration.
What Is IL-1β (IL-1b)? Function, Signaling, and Biological Role
An overview of IL-1β, including its signaling pathways, involvement in disease mechanisms, and potential therapeutic targets.
Mitochondrial Dysfunction in Microglia & Astrocytes
The role of mitochondrial dysfunction in microglia and astrocytes in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and ALS.
Microglia, Astrocytes & α-Synuclein in Parkinson’s Disease
How α-synuclein influences microglia and astrocytes in Parkinson’s disease and other synucleinopathies.
Interleukin-1 Beta (IL-1β) and Neurodegenerative Diseases
The role of IL-1beta in neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS).
Microglia, Astrocytes & Tau in Neurodegenerative Diseases
How glial-driven neuroinflammation fuels tau aggregation, propagation, and neuronal loss in Alzheimer’s disease and other tauopathies.