中枢神経系(CNS)におけるTNF-αの役割とは何でしょうか?
腫瘍壊死因子-α(TNF-α)は炎症性サイトカインの一種で、炎症や免疫反応の調節に重要な役割を果たしています。中枢神経系(CNS)においては、TNF-αには二つの機能があり、健康な状態では有益に作用しますが、病的な状態では調節不全により有害な影響を及ぼす可能性があります。恒常性維持の状態では、TNF-αは神経可塑性、ミエリン形成、組織修復をサポートします。しかし、そのレベルが上昇すると、興奮毒性、慢性炎症、血液脳関門(BBB)の破壊などの有害な影響を引き起こす可能性があります。TNF-αは可溶性と膜貫通型の両方の形態で存在し、主に2つの受容体を介して作用します。TNF受容体タイプ1(TNFR1)およびTNF受容体タイプ2(TNFR2)です。その発現は、特に活性化B細胞核因子(NF-κB)などのさまざまな転写因子によって厳密に制御されています。NF-κBが活性化されると、TNF-αの産生を促進するだけでなく、IL-6やIL-1βなどの他の炎症促進性サイトカインの発現も引き起こします。 TNF-αとNF-κBの相互作用は正のフィードバックループを生み出し、炎症反応を増幅し、免疫反応を持続させます(Gonzalez Caldito, 2023)。
神経炎症時には、中枢神経系の常在免疫細胞であるミクログリアがTNF-αの主な供給源となりますが、アストロサイトの放出も寄与します(Olmos, 2014)。 神経変性疾患では、ミクログリアの反応性、ミクログリアの老化や老化関連分泌表現型(SASP)などの他の状態が、TNF-αや他の炎症促進性サイトカインの産生増加につながり、慢性炎症環境が持続します。炎症性サイトカインであるインターフェロンガンマ(IFN-γ)などは、さらにミクログリアを刺激してTNF-αを放出させ、炎症を悪化させる可能性があります。この進行中の炎症反応は、アルツハイマー病(AD)、パーキンソン病(PD)、筋萎縮性側索硬化症(ALS)などの神経変性疾患の進行を加速させます。
TNF-αを標的とした治療戦略は、中枢神経系の自己免疫疾患の治療に有望であることが示されており、神経変性疾患の神経病理学的特徴を修正する方法を提供する可能性があります(Torres-Acosta, 2020;Gonzalez Caldito, 2023)。しかし、抗TNF-α療法は炎症を軽減することができますが、その効果は複雑であり、多発性硬化症(MS)のような病状を悪化させるなど、時には予期せぬ結果を招くこともあります(Fresegna, 2020)。これらの副作用は、TNF-αとその受容体が文脈に応じて炎症促進効果と保護効果の両方を持つという二重の役割を持つことが原因である可能性があります。さらに、特に疾患の進行の早期と後期におけるTNF-αの時間的制御を研究することで、TNF-αの調節のタイミングが治療結果にどのような影響を与えるかについての洞察が得られる可能性があります(Guidotti, 2021)。その結果、治療戦略を改善するためには、TNF-αの中枢神経系における役割をより深く理解し、これらの治療法の安全性と有効性に関するさらなる研究が必要となります。
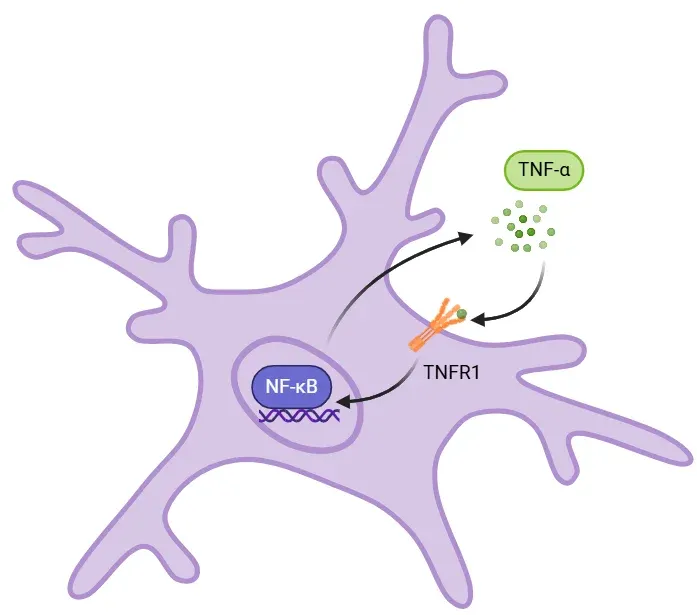
可溶性TNF-αがTNFR1に結合すると、NF-κBを活性化するシグナル伝達カスケード(図示せず)が開始されます。核内でNF-κBは、IL-6、IL-1β、TNF-α自身を含む炎症性サイトカインの転写を促進します。このサイトカインのアップレギュレーションは、正のフィードバックループを通じて免疫反応を増幅します。TNF-αがNF-κB活性を高め、それがさらにTNF-αの産生を促進します。TNF-αレベルの上昇は、組織損傷、興奮毒性、その他の有害な影響を引き起こす可能性があります。神経変性疾患など、制御不能な場合、このループは慢性的な炎症を維持します。
TNF-αは、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症などの神経変性疾患とどのように関連しているのでしょうか?
アルツハイマー病(AD)
ADは、進行性の認知機能低下、記憶障害、アミロイドベータ(Aβ)プラークおよびタウ神経原線維変化の蓄積を特徴とする神経変性疾患です。 炎症性サイトカインがADの病態生理において重要な役割を果たしていることを示す証拠が増えています。TNF-α、IL-1β、IL-6のレベル上昇が、アルツハイマー病患者の血液および脳脊髄液(CSF)で検出されています(Brosseron, 2014)。しかし、病気の段階の違いが原因で、相反する結果を報告している研究もあります。こうした相違はあるものの、TNF-αのようなサイトカインは、アルツハイマー病の進行に伴い増加する傾向にあります。ただし、そのレベルは信頼性の高いバイオマーカーとして機能するには低すぎる場合が多いです。サイトカインのレベルと疾患の進行の正確な関係は、現在も研究が進められている分野です。一部の研究では、サイトカインのレベル上昇はアルツハイマー病発症リスクの増加を示唆する可能性があり、サイトカインとそれぞれの受容体の比率は、個々のサイトカインのレベルよりも正確な診断マーカーとなる可能性があることが示唆されています(Brosseron, 2014)。
ミクログリアの機能不全もまた、アルツハイマー病の病態における重要な要因です。βアミロイド蓄積のモデルでは、老化したミクログリアは貪食活性の低下と、TNF-αやIL-1βなどの炎症性サイトカインの発現増加を示します(Hickman, 2008)。この機能不全が引き金となってフィードバックループが形成され、アミロイドβプラークの蓄積がミクログリアを活性化し、TNF-αの放出を引き起こします。このサイトカインの放出は神経炎症を増幅し、神経細胞の損傷を加速させます。持続的なサイトカインの放出はさらなる神経変性を促し、それがまた炎症反応を悪化させ、アミロイドβの蓄積を増加させます。ミクログリアがアミロイドβプラークを効果的に除去できないため、有毒な凝集体が蓄積されます。このプラークの蓄積、ミクログリアの反応性、TNF-αの放出、神経細胞の損傷というサイクルが神経変性プロセスを永続させ、アルツハイマー病の進行を加速させます(Hickman, 2008)。
アルツハイマー病におけるTNF-α阻害の治療効果を調査する臨床試験では、結果はまちまちです。小規模な非盲検試験では、TNF-α阻害剤エタネルセプトを脊髄周辺経路で投与したところ、認知と行動の改善が見られ、有望な結果が示されました(Tobinick, 2008)。しかし、同じ治療を末梢で投与したところ、これらの領域では有意な変化は観察されませんでした(Butchart, 2015)。しかし、可溶性TNF-αを選択的に標的とするTNF-α阻害剤であるXPro1595の第1b相試験では、アルツハイマー病において有望な結果が示されています(NCT03943264)。この新しいアプローチは、可溶性のTNF-αに焦点を当てており、アルツハイマー病に関連する炎症反応を調節する、より的を絞った効果的な方法を提供できる可能性があります。
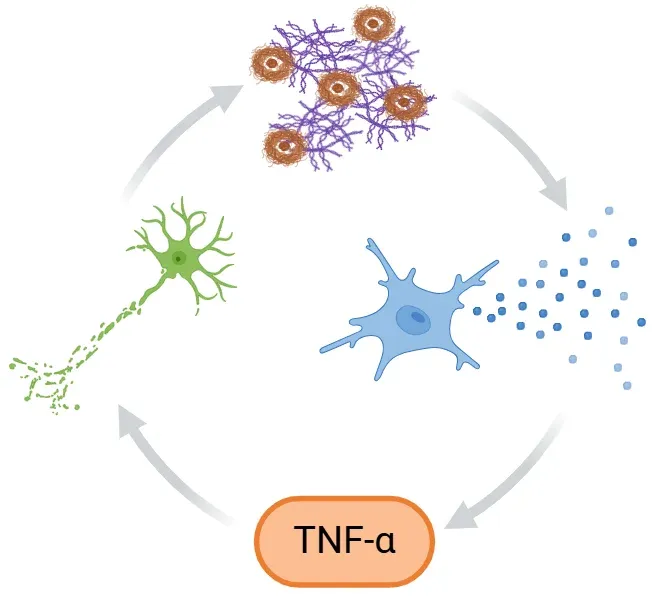
アルツハイマー病(AD)における正のフィードバックループの提案。ここでは、Aβプラークとタウタンパク質の凝集がミクログリアを活性化し、TNF-αなどの炎症促進性サイトカインが放出されます。この放出により神経炎症と神経変性が増幅され、ミクログリアの除去機能が損なわれることで有毒なタンパク質の蓄積が増加し、さらにサイクルが悪化してADの進行が加速します。
パーキンソン病(PD)
PDは、黒質緻密部(SNpc)のドパミン作動性ニューロンの減少を特徴とする進行性の神経変性疾患であり、振戦、筋固縮、動作緩慢などの運動症状を引き起こします。 これらの特徴的な運動症状に加え、PDは認知機能低下、睡眠障害、うつ病などの非運動症状も引き起こします。
慢性神経炎症は、パーキンソン病の病態生理における重要な因子であることが確認されています。パーキンソン病患者の死後脳におけるSNの分析では、ミクログリアの反応性とTNF-αなどの炎症促進性サイトカインのレベル上昇が明らかになっています。これらのサイトカインは、パーキンソン病患者の血清にも認められます(Collins, 2012)。TNFR1はヒトのSNpcのドーパミン作動性神経細胞に発現しており、TNF-αが病気の進行に関与していることをさらに示唆しています。さらに、パーキンソン病の動物モデルでは、血清および脳脊髄液の両方でTNF-αの発現が増加しています(Collins, 2012)。
炎症性サイトカインをコードする遺伝子を制御する転写因子NF-κBの活性化もまた、パーキンソン病患者で上昇しており、炎症経路がこの疾患とさらに結びついています(Mogi, 2007)。研究により、慢性的に反応しているミクログリアは、TNF-αなどの炎症促進性サイトカインを大量に放出することが明らかになっています。このサイトカインは、ドーパミン作動性ニューロンを損傷するだけでなく、PDにおけるミクログリアのさらなる反応性、神経炎症、神経変性を引き起こすフィードバックループを永続化させることも示されています(Collins, 2012)。
動物モデルを用いた研究により、TNF-αを遮断することでパーキンソン病における神経変性を大幅に軽減できることが実証されています。例えば、TNF-α合成阻害剤であるサリドマイドによる治療はMPTP誘発性のドーパミン減少を防ぎ、TNF-α欠損マウスでは線条体のドーパミン減少と死亡率が大幅に減少することが示されています(Ferger, 2004)。さらに、TNF-α阻害剤であるXENP345の使用は、マウスのパーキンソン病モデルにおいてドーパミン作動性神経細胞の死を減少させることが示されています(McCoy, 2006)。これらの知見は、TNF-αを標的とすることが、パーキンソン病における炎症プロセスを調節し、病気の進行を遅らせるための有望な治療アプローチとなり得ることを示唆しています。
筋萎縮性側索硬化症(ALS)
ALSは主に運動ニューロンを標的とする進行性の神経変性疾患であり、筋力低下、発声や嚥下の困難、最終的には痙性や麻痺を引き起こします。これらの主な症状に加え、神経炎症は疾患の進行に重要な役割を果たしています。ALS患者の血漿および血清、ならびにALSの動物モデルにおいて、TNF-αのレベル上昇が検出されています(Olmos, 2014;Vu, 2017)。さらに、NF-κBのアップレギュレーションは、ALS患者と、ALS研究で一般的に使用されるモデルであるスーパーオキシドジスムターゼ1(SOD1)トランスジェニックマウスモデルの両方の脊髄ミクログリアで観察されています(Frakes, 2014)。これらの知見は、ALSの病態生理におけるTNF-αの関与を裏付けるものです。
ALSにおけるTNF-αの役割をより深く理解するために、特にSOD1トランスジェニックマウスモデルにおいて遺伝子ノックアウト研究が行われてきました。これらの研究では、TNF-αの遺伝子を削除しても、寿命や運動ニューロン変性への著しい影響は見られませんでした(Gowing, 2006)。これらの結果は、TNF-αがALSで観察される炎症プロセスに寄与している可能性はあるものの、特にSOD1変異の文脈においては、運動ニューロン変性の直接的な原因ではないかもしれないことを示唆しています。
一方、サリドマイドやレナリドミドのようなTNF-α阻害剤を用いた研究では、有望な結果が示されています。SOD1トランスジェニックマウスにおいて、これらの阻害剤は運動機能を改善し、運動ニューロン細胞死を減少させました(Kiaei, 2006)。これらの結果は、TNF-αを標的とすることがALSの有効な治療アプローチとなり得ることを示唆しています。しかし、ALS患者を対象にサリドマイドの使用を調査した第2相臨床試験では、これらの有望な結果は再現されませんでした。サリドマイドの日常的な投与は有益な効果を示さず、いくつかの有害な副作用が認められました(Stommel, 2009)。
これらの相反する結果は、治療のタイミングによるものかもしれません。TNF-α阻害の効果は、ALSの進行段階によって異なる可能性があるため、ALSにおけるTNF-αの複雑な役割をより深く理解し、治療戦略を改善するためのさらなる研究の必要性が浮き彫りになっています(Guidotti, 2021)。TNF-α阻害は依然として治療の可能性を秘めた手段ではありますが、ALSにおけるこの炎症経路を標的とする最も効果的な方法を決定するには、さらなる研究が不可欠です。
結論として、TNF-αは、アルツハイマー病、パーキンソン病、ALSなどの疾患にわたる神経炎症および神経変性において中心的な役割を果たしています。その根本的なメカニズムには違いがあるものの、TNF-αが引き起こす炎症プロセスは、疾患の進行における共通因子であるようです。TNF-αを標的とした治療は前臨床モデルでは有望な結果を示していますが、臨床結果は一貫しておらず、TNF-αの役割をより深く理解する必要性を浮き彫りにしています。今後の研究では、TNF-αのシグナル伝達を調節する標的治療の改良に焦点を当てるべきであり、それにより、これらの神経変性疾患の進行を遅らせたり、食い止めたりする治療法が提供される可能性もあります。
TNF-αを調節することが神経変性疾患の進行を遅らせたり、予防したりする可能性があるという根拠は何でしょうか?
神経炎症の進行において重要な役割を果たすTNF-αは、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症など、多くの神経変性疾患の進行の中心的な役割を担っています。TNF-αが中枢神経系で慢性的に増加すると、神経細胞の損傷と有毒タンパク質の蓄積が起こり、これが疾患の進行に寄与します。そのため、TNF-αを調節することは、これらの疾患の進行を遅らせる、あるいは停止させるための有望な戦略として浮上しています。
自己免疫疾患や炎症性疾患の治療に有効な抗TNF-α療法は、神経変性疾患への応用も期待されています(Gonzalez Caldito, 2023)。エタネルセプト(エンブレル®)やインフリキシマブ(レミケード®)などの薬剤は、関節リウマチやその他の末梢性炎症性疾患の治療に広く使用されていますが、血液脳関門の透過性が低いため、中枢神経系疾患に対する効果は限定的です(Decourt, 2017)。アダリムマブ(ヒュミラ®)、ゴリムマブ(シムポニー®)、セルトリズマブ ペゴル(シムジア®)などの他のTNF-α阻害剤も、中枢神経系へのアクセスという点で同様の課題に直面しています。
このCNS疾患におけるTNF-α阻害の課題は、感染症や脱髄疾患に対する感受性が高まるなど、深刻な副作用のリスクによってさらに複雑化しています(Kemanetzoglou, 2017)。例えば、MSでは、レナリドミドのような治療法は病気の経過を悪化させています(Maguire, 2021)。 神経保護と炎症におけるTNF-αの二重の役割は、より選択的な標的設定の必要性を強調しています。 TNFR1とTNFR2の研究により、TNFR2を温存しながらTNFR1を選択的に標的とすることが、MSの治療に、より安全で効果的なアプローチをもたらす可能性があることが強調されています(Fresegna, 2020)。
これらの限界に対応して、CNS用途にTNFR1を特異的に標的とする選択的TNF-α阻害剤、例えばアトロサブやアトロシムマブなどの開発は、実験的自己免疫性脳脊髄炎(EAE)などの神経炎症の非臨床モデルにおいて有望な結果を示しています(リヒター、2021年)。同様に、可溶性TNF-αに選択的なTNF-α阻害剤であるXPro1595の第1b相試験では、アルツハイマー病において有望な結果が示されています(NCT03943264)。これらの治療法は、神経変性疾患の安全性と有効性を改善し、より標的を絞ったアプローチを提供することを目的としています。
前臨床試験の結果は有望ですが、ヒトにおける選択的TNF-α阻害剤の安全性と有効性を確立するには、さらなる臨床試験が必要です。これらの研究は、これらの治療法が神経変性疾患患者に恩恵をもたらすことができるかどうかを判断する上で極めて重要となります。研究が継続されるにつれ、中枢神経系に特化したTNF-α療法の開発は、神経変性を遅らせたり予防したり、患者の予後を改善したり、神経炎症治療の分野を発展させたりするための重要なアプローチとなる可能性があります(Decourt, 2017;Hampel, 2020;Zahedipour, 2022)。
私たちのチームは、ミクログリアにおけるTNF-αの機能や神経変性疾患への関与についてのご質問、あるいは治療効果の研究に使用しているAD、ALS、PDモデルに関する特定の情報提供など、どのようなご質問にも喜んでお答えいたします。
神経変性疾患モデルについてさらに詳しく知る
関連コンテンツ
神経変性疾患におけるTNF-αとミクログリアに関する最新情報、および神経変性疾患の動物モデルにおける治療薬の評価に関するベストプラクティス。
神経変性疾患におけるTNF-αとアストロサイト
アストロサイトにおけるTNF-αシグナル伝達の概要、神経変性におけるその役割、およびこの経路を標的とする治療戦略について。
神経変性疾患におけるミクログリア、アストロサイト、およびタウ
グリア細胞による神経炎症が、アルツハイマー病およびその他のタウオパチーにおいて、タウタンパク質の凝集、伝播、および神経細胞の喪失を促進するメカニズム。
パーキンソン病におけるミクログリア、アストロサイト、およびα-シヌクレイン
α-シヌクレインがパーキンソン病およびその他のシヌクレイン病においてミクログリアおよびアストロサイトに及ぼす影響。
ミトコンドリア機能障害とミクログリアおよびアストロサイト
アルツハイマー病、パーキンソン病、ALSを含む神経変性疾患におけるミトコンドリア機能障害のミクログリアおよびアストロサイトにおける役割。
ミクログリアの老化と神経変性疾患
本リソースでは、アルツハイマー病(AD)やパーキンソン病(PD)などの神経変性疾患におけるミクログリアの老化とその役割について概説します。
ALS, アルツハイマー病, パーキンソン病におけるミクログリアの形態
ミクログリアの形態解析の概要と, 神経変性疾患の研究および創薬・薬剤開発への応用。
NLRP3インフラマソームと神経変性疾患
NLRP3インフラマソームの概要と、アルツハイマー病、パーキンソン病、ALSなどの神経変性疾患におけるその役割について。