Microglie, astrocytes et protéine tau dans les maladies neurodégénératives
Comment la neuroinflammation induite par les cellules gliales favorise l'agrégation et la propagation de la protéine tau ainsi que la perte neuronale dans la maladie d'Alzheimer et d'autres tauopathies.
Cette ressource décrit :
- Comment les microglies et les astrocytes interagissent-ils dans les maladies neurodégénératives ?
- Comment la neuroinflammation est-elle liée aux tauopathies ?
- Comment les microglies contribuent-elles à la pathologie tau dans les maladies neurodégénératives ?
- Comment les astrocytes amplifient-ils et transmettent-ils la pathologie tau ?
- La clairance des agrégats de tau médiée par les cellules gliales peut-elle être améliorée par des moyens thérapeutiques ?
Comment les microglies et les astrocytes interagissent-ils dans les maladies neurodégénératives?
Les microglies et les astrocytes sont deux types clés de cellules gliales qui jouent un rôle essentiel dans le maintien de l'homéostasie et la réponse aux lésions au sein du système nerveux central (SNC). Ces deux types de cellules contribuent à la réponse immunitaire du cerveau et jouent un rôle crucial dans la neuroinflammation, une caractéristique commune aux maladies neurodégénératives.
Microglies
Les microglies sont les principales cellules immunitaires du SNC. Elles sont dérivées des macrophages du sac vitellin et se distinguent des cellules immunitaires périphériques (Ginhoux, 2010; Leavy, 2010). Elles sont très dynamiques et surveillent en permanence le microenvironnement du cerveau à l'aide de leur répertoire de récepteurs spécialisés (Qin, 2023). Les microglies jouent un rôle crucial dans la phagocytose, l'élagage synaptique et la sécrétion de molécules de signalisation qui régulent les fonctions neuronales (Gao, 2023).
Des avancées récentes ont révélé un large éventail d'états microgliaux, allant au-delà de la classification binaire traditionnelle des microglies M1 (pro-inflammatoires) et M2 (anti-inflammatoires). Au contraire, les microglies présentent un spectre de phénotypes fonctionnels qui varient en fonction des signaux environnementaux, des facteurs génétiques et de la progression de la maladie. Par exemple, dans la maladie d'Alzheimer (MA), les microglies associées à la maladie (DAM) sont localisées près des plaques amyloïdes et contribuent à l'élimination des agrégats amyloïdes (Keren-Shaul, 2017; Gao, 2023). À l'inverse, l'activation prolongée des microglies peut entraîner une production excessive de cytokines, altérant la fonction synaptique et exacerbant les lésions neuronales (Kwon, 2020). Les microglies libèrent également des vésicules extracellulaires qui contribuent à la neurodégénérescence en propageant des protéines mal repliées, telles que la protéine tau et la protéine bêta-amyloïde dans la MA et l'alpha-synucléine dans la maladie de Parkinson (MP) (Gao, 2023).
Astrocytes
Les astrocytes sont les cellules neuronales les plus abondantes dans le SNC. Ils jouent un rôle essentiel dans le maintien de l'homéostasie, la régulation du flux sanguin et le soutien de la transmission synaptique. Ils participent activement à la modulation de la barrière hémato-encéphalique (BHE) et sont un élément clé du système glymphatique. Ils jouent un rôle majeur dans la fonction synaptique et neuronale, dans le contrôle de l'équilibre extracellulaire des ions et des neurotransmetteurs et dans l'élimination des cellules mortes (Abbott, 2010; Sweeney, 2018; Kwon, 2020).
Tout comme les microglies, les astrocytes peuvent exister dans des états réactifs allant de protecteurs à nocifs. Les astrocytes pro-inflammatoires (phénotype A1) libèrent des cytokines, telles que l'IL-1β, le TNF-α et les protéines de la cascade du complément, qui sont connues pour endommager les neurones et les synapses. À l'inverse, les astrocytes anti-inflammatoires (phénotype A2) favorisent la survie et la réparation des neurones en sécrétant des facteurs neurotrophiques et d'autres molécules protectrices. Le dysfonctionnement astrocytaire est couramment observé dans les maladies neurodégénératives, où il exacerbe souvent la neuroinflammation et contribue à la progression de la maladie (Kwon, 2020).
Dans la MA, par exemple, l'activation astrocytaire est associée à une altération de la clairance du glutamate, entraînant une excitotoxicité et la mort neuronale. De plus, les astrocytes réactifs induits par les cytokines microgliales peuvent compromettre la BHE, aggravant encore la vulnérabilité neuronale (Kwon, 2020). De même, dans la SLA, les astrocytes réactifs exprimant des facteurs toxiques, tels que le C3, contribuent à la dégénérescence des motoneurones (Gao, 2023).
Les microglies et les astrocytes
Les microglies et les astrocytes sont essentiels au maintien de l'homéostasie du SNC et jouent un rôle crucial dans la réponse aux lésions et aux maladies. Leurs interactions sont très dynamiques et peuvent influencer la neuroinflammation et la neurodégénérescence de manière protectrice ou néfaste, selon leur état et le contexte environnemental (Liu, 2020; Kim, 2021; Gotoh, 2023). En réponse à des stimuli pathologiques, les microglies s'activent et libèrent des facteurs pro-inflammatoires, tels que l'IL-1α, le TNF-α et le C1q. Ces molécules poussent les astrocytes vers un phénotype A1 réactif, caractérisé par la libération de cytokines pro-inflammatoires supplémentaires qui maintiennent l'activation microgliale. Cette signalisation réciproque établit une boucle de rétroaction néfaste qui amplifie l'inflammation et accélère les processus neurodégénératifs (Gao, 2023). Dans cet état réactif, les microglies augmentent également l'élagage synaptique, un processus normalement essentiel au raffinement des circuits neuronaux, mais qui peut devenir nocif dans des conditions inflammatoires. Parallèlement, les astrocytes stimulent les neurones pour qu'ils libèrent des protéines du complément, qui activent davantage les microglies pour qu'elles phagocytent les synapses et même des neurones entiers, contribuant ainsi à la perte neuronale (Sierra, 2013). Ce cycle inflammatoire croissant peut perturber la barrière hémato-encéphalique (BHE), affaiblissant sa fonction protectrice et permettant aux cellules immunitaires périphériques, telles que les lymphocytes B, les lymphocytes T et les monocytes, de s'infiltrer dans le SNC. Ces cellules infiltrées exacerbent l'inflammation en augmentant la libération de cytokines par les cellules gliales, créant un environnement d'activation immunitaire soutenue qui peut endommager davantage les tissus neuronaux (Abbott, 2010; Sweeney, 2018).
Malgré ces interactions néfastes, les microglies et les astrocytes sont également capables de favoriser des processus protecteurs et régénérateurs. Dans certaines conditions, les microglies peuvent induire les astrocytes à adopter le phénotype anti-inflammatoire A2 par le biais de la signalisation IL-10, favorisant ainsi un environnement plus réparateur qui limite les lésions tissulaires et favorise la réparation (Mohammad, 2024). À la suite d'une lésion du SNC, les astrocytes augmentent leur prolifération et forment une « cicatrice gliale », une barrière physique qui aide à contenir le site de la lésion et à prévenir d'autres lésions tissulaires. Bien que cette cicatrice puisse parfois inhiber la régénération neuronale, elle joue un rôle essentiel dans la stabilisation de l'environnement du SNC au cours des premières étapes de la récupération. Au-delà de la formation de cicatrices, les microglies et les astrocytes libèrent des facteurs qui favorisent la reconstruction de la matrice extracellulaire (MEC), la néovascularisation et l'angiogenèse. Ces processus créent un environnement favorable à la survie des neurones, favorisent la migration des cellules souches pour remplacer les neurones endommagés et facilitent le remodelage axonal afin de restaurer les connexions perdues (Yang, 2019; Heithoff, 2021).
En fin de compte, les interactions entre les microglies et les astrocytes sont complexes et étroitement régulées, avec des résultats qui peuvent varier considérablement en fonction de l'équilibre entre les signaux pro-inflammatoires et anti-inflammatoires. Si leur interaction peut entraîner une inflammation néfaste et une perte neuronale, elle est tout aussi essentielle pour favoriser la réparation et le rétablissement de l'homéostasie du SNC après une lésion ou une maladie. Il est essentiel de comprendre ces deux rôles pour élaborer des stratégies visant à atténuer les lésions neurodégénératives tout en renforçant la capacité de régénération intrinsèque du SNC.
Quel est le lien entre la neuroinflammation et les tauopathies?
Les tauopathies sont un groupe de troubles neurodégénératifs caractérisés par l'accumulation progressive de protéines tau hyperphosphorylées dans des agrégats fibrillaires intracellulaires. Ces agrégats, qui varient en termes de composition isoforme, de conformation structurelle et de localisation, se déposent le plus souvent dans les neurones. Les tauopathies sont classées en formes primaires, où le dépôt de tau est la principale caractéristique pathologique, par exemple la paralysie supranucléaire progressive (PSP), la dégénérescence corticobasale (CBD) et la démence frontotemporale (FTD); les formes secondaires, qui présentent des contributions pathologiques supplémentaires, telles que la bêta-amyloïde dans la maladie d'Alzheimer (MA); et les formes géographiques, comme le parkinsonisme guadeloupéen (Lannuzel, 2007). Au cours de la progression de la maladie, les protéines tau subissent des modifications post-traductionnelles qui les font se replier en agrégats riches en feuillets bêta, qui se propagent ensuite de leur site de dépôt d'origine vers des régions anatomiquement connectées par un mécanisme de type prion. Ce processus implique des graines protéopathiques tau qui recrutent des monomères tau (Jucker, 2018). Alors que les cellules gliales, notamment les microglies et les astrocytes, tentent initialement d'atténuer les dommages en phagocytant la protéine tau extracellulaire et en libérant des facteurs protecteurs, une exposition chronique les conduit à des états réactifs. Ces états réactifs se caractérisent par l'activation des voies NF-κB, la transcription induite par l'inflammasome, la libération excessive de cytokines, l'altération de la clairance lysosomale et des erreurs dans l'élagage synaptique. Ces conditions neuroinflammatoires persistantes favorisent la formation de germes tau, accélèrent le dysfonctionnement neuronal et sont étroitement corrélées au déclin cognitif, aggravant encore la progression des tauopathies.
Comment les microglies contribuent-elles à la pathologie tau dans les maladies neurodégénératives?
Il est de plus en plus admis que les microglies pro-inflammatoires et hypofonctionnelles jouent des rôles distincts dans la pathologie tau et la neurodégénérescence (Streit, 2014; Angelova, 2019; Odfalk, 2022). Odfalk a très succinctement identifié ce paradigme dans une récente revue intitulée «Microglia: Friend and foe in tauopathy» (Odfalk, 2022). Au cours des premiers stades de la maladie, les microglies jouent un rôle protecteur. Ils migrent vers les neurites dystrophiques, engloutissent la protéine tau soluble et filamenteuse libérée par les neurones mourants et sécrètent des médiateurs anti-inflammatoires qui aident à rétablir l'homéostasie tissulaire. Cependant, cet état de surveillance bénéfique est fragile. Un stress protéopathique persistant, une sensibilisation liée à l'âge, des allèles génétiques à risque et/ou des atteintes cérébrovasculaires comorbides conduisent les microglies vers un phénotype associé à la maladie (DAM). Les DAM régulent à la baisse les marqueurs homéostatiques, tout en régulant à la hausse les gènes phagolysosomaux et liés au métabolisme lipidique qui, paradoxalement, favorisent l'absorption, la fragmentation intracellulaire et la libération ultérieure d'espèces de tau plus aptes à se propager.
Il existe des preuves substantielles du rôle positif de la phagocytose des microglies dans l'élimination de la protéine tau (Luo, 2015; Bolós, 2017; Leyns, 2017). Il a été démontré expérimentalement que la pathologie tau s'améliore lorsque la neuroinflammation est réduite en modifiant le phénotype des microglies, soit par la suppression du TNFα (Gabbita, 2015), soit par le blocage de la signalisation de l'IL-1 chez des souris 3xTgAD (Kitazawa, 2011). Il existe également des preuves que les microglies contribuent directement à la progression de la pathologie tau par divers mécanismes, notamment la dérégulation de leurs fonctions homéostatiques (Perea, 2018; 2020). Par exemple, les microglies peuvent faciliter la transmission intercellulaire des germes tau qui forment ensuite des agrégats. Ce mécanisme est corroboré par Asai et al., qui ont démontré que la déplétion des microglies réduisait la propagation de la protéine tau (Asai, 2015). De plus, il existe des preuves convaincantes que les microglies associées aux plaques favorisent la propagation de la protéine tau dans des modèles murins transgéniques (Clayton, 2021).
Le rôle du récepteur de la fractalkine CX3CR1 (Perea, 2018) est un exemple illustratif de l'équilibre normal et pathologique qui existe dans le contexte des tauopathies. Dans des conditions normales, les microglies restent au repos, où le ligand CX3C (CX3CL1) exprimé sur les neurones (Jiang, 2023) et le récepteur (CX3CR1) exprimé sur les microglies (Bhaskar, 2010; Zhan, 2014; Maphis, 2015) interagissent pour maintenir une fonctionnalité normale. Cet axe est maintenu par la liaison des formes membranaires et solubles de CX3CL1 au récepteur microglial CX3CR1, ce qui garantit le bon fonctionnement du système immunitaire. En présence de tau extracellulaire, les microglies s'activent et tentent d'éliminer l'excès de tau par phagocytose, un processus influencé par l'axe CX3CL1/CX3CR1. Dans la maladie d'Alzheimer, à mesure que la pathologie progresse et que la mort neuronale augmente, on observe une accumulation notable de tau extracellulaire, qui interfère avec le fonctionnement normal de la voie CX3CL1/CX3CR1. L'excès de tau entre en compétition avec CX3CL1 pour se lier à CX3CR1, ce qui altère l'élimination de la tau par les microglies (Bolós, 2017). En réponse, les microglies deviennent hyperactivées, prolifèrent et tentent de compenser en produisant davantage de CX3CL1 et de CX3CR1. Malgré cette augmentation, l'axe CX3CL1/CX3CR1 se dérégule, contribuant ainsi davantage à l'accumulation de tau et à la progression de la maladie. Cette réponse microgliale dysfonctionnelle met en évidence le rôle complexe de la régulation immunitaire.
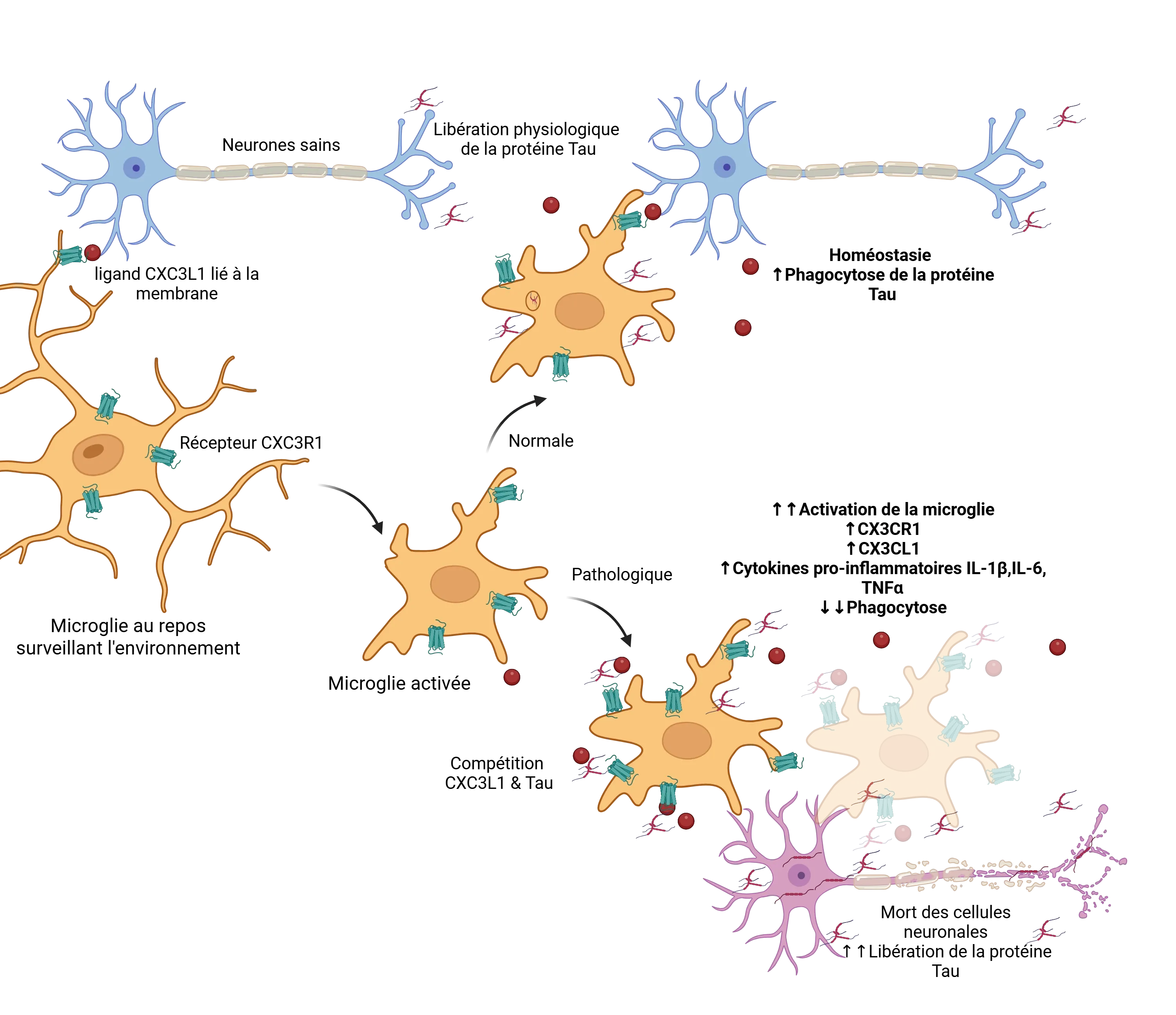
Des signaux tels que CXCL1/CXC3R1 contribuent à maintenir les microglies à l'état de repos pendant qu'elles surveillent leur environnement. En présence de tau extracellulaire, ces microglies sont activées et, dans des conditions normales, phagocytent la tau et contribuent à maintenir l'homéostasie. Dans des conditions pathologiques, l'excès de tau entre en compétition avec les interactions CXCL1/CXC3R1, poussant les microglies activées dans un état dysfonctionnel, avec une activation accrue, une diminution de la phagocytose, une réduction de la clairance de la protéine tau et, par conséquent, une augmentation de l'accumulation extracellulaire de tau, ce qui exacerbe le cycle toxique. Figure reproduite à partir de Perea et al. (Perea, 2018) sous licence Creative Commons Attribution.
Comment les astrocytes amplifient-ils et transmettent-ils la pathologie tau?
Les astrocytes sont de plus en plus reconnus comme des acteurs centraux de la pathologie tau, non seulement en tant que composants passifs affectés par le dysfonctionnement neuronal, mais aussi en tant que contributeurs actifs à la régulation de l'homéostasie tau et à sa propagation pathologique dans les régions du cerveau. Des recherches récentes indiquent que les astrocytes participent à des mécanismes complexes d'internalisation de la protéine tau, influençant ainsi son accumulation et son élimination. Ces cellules gliales jouent un double rôle: d'une part, elles peuvent agir comme des tampons en tentant d'atténuer la charge extracellulaire de la protéine tau, tandis que d'autre part, leur dysfonctionnement ou leur hyperactivation peut amplifier l'agrégation et la propagation de la protéine tau. Ce rôle nuancé met en évidence les astrocytes comme cibles potentielles pour des interventions thérapeutiques visant à moduler la dynamique de la protéine tau dans les maladies neurodégénératives (Reid, 2020).
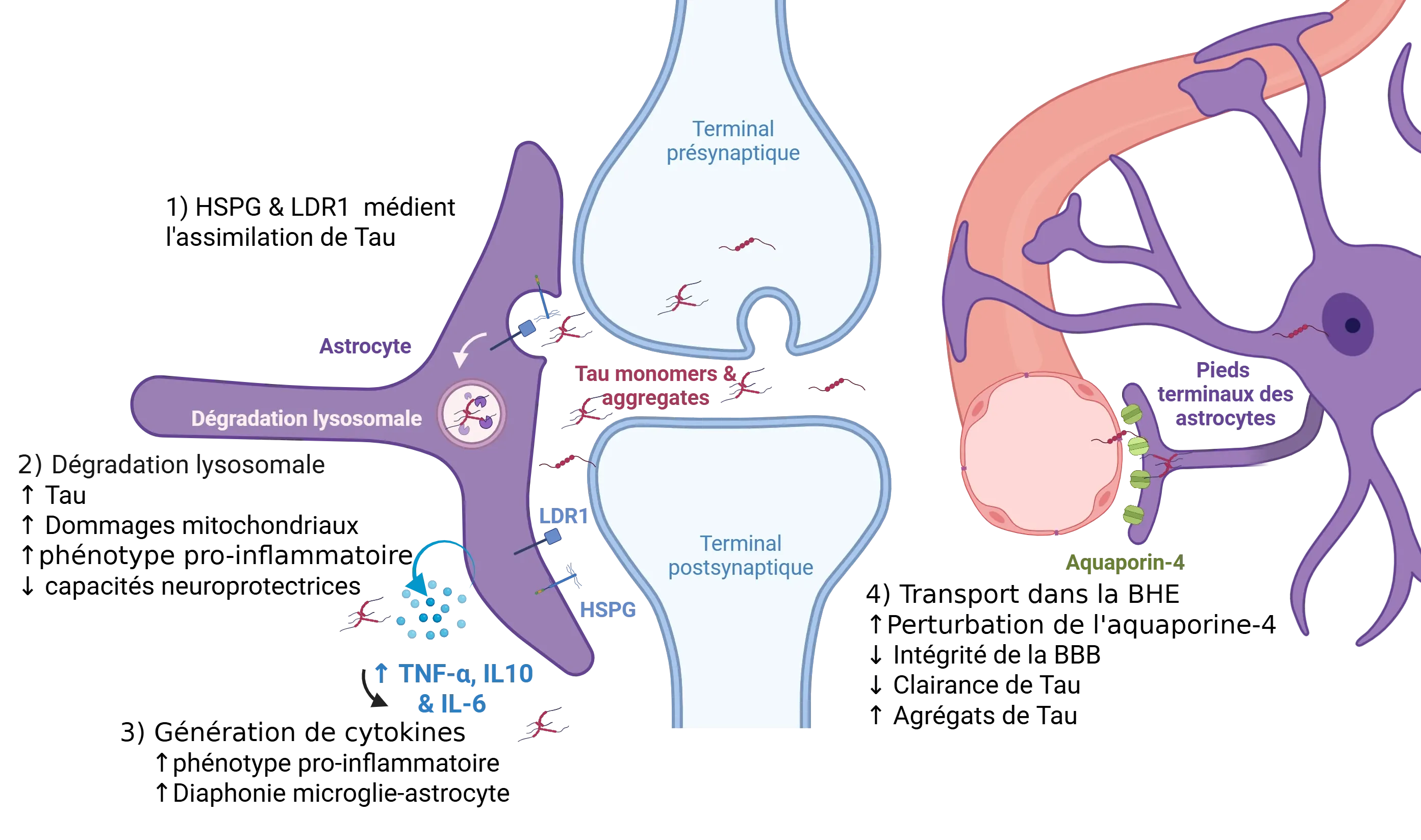
Les astrocytes jouent un rôle actif dans la modulation de la propagation de la pathologie tau au sein du système nerveux central. Les neurones libèrent des espèces tau dans l'espace extracellulaire par plusieurs voies, notamment la sécrétion vésiculaire et la libération synaptique au niveau de la présynapse. Les astrocytes périsynaptiques internalisent la protéine tau extracellulaire par endocytose médiée par des récepteurs, principalement par l'intermédiaire de protéoglycanes spécifiques de l'héparane sulfate (HSPG), qui se lient préférentiellement à la protéine tau agrégée, et de la protéine 1 apparentée au récepteur des lipoprotéines de basse densité (LRP1), qui intervient dans l'absorption de la protéine tau monomérique et oligomérique (1). La perturbation de ces voies peut modifier l'équilibre entre la clairance et l'accumulation de la protéine tau extracellulaire, ce qui peut favoriser sa propagation. Une fois internalisée, la protéine tau est acheminée vers les lysosomes pour y être dégradée (2) ; cependant, une altération de la fonction lysosomale, due à une surcharge intrinsèque ou à une signalisation pathologique, entraîne une accumulation intracellulaire de la protéine tau et la formation d'agrégats capables de se propager. Parallèlement, un milieu pro-inflammatoire, caractérisé par des taux élevés de cytokines, telles que le facteur de nécrose tumorale alpha (TNF-α) et les interleukines IL-10 et IL-6, amplifie la réactivité astrocytaire et facilite la communication entre les astrocytes et les microglies (3), augmentant ainsi le pool de tau pathogènes disponibles pour la propagation. De plus, la perturbation de la localisation de l'aquaporine-4 (AQP4) au niveau des pieds terminaux des astrocytes périvasculaires compromet la clairance glymphatique de la protéine tau (4). La perturbation de ce mécanisme de clairance périvasculaire entraîne une accumulation progressive de tau extracellulaire, exacerbant ainsi la propagation de la pathologie tau dans tout le parenchyme cérébral. Figure tirée de Reid et al. (Reid, 2020) sous licence Creative Commons Attribution.
Au niveau de la synapse, les neurones libèrent à la fois des protéines tau monomériques et agrégées autour de la fente synaptique, où les astrocytes les capturent rapidement par endocytose médiée par des récepteurs (Reid, 2020). Les astrocytes sont dotés de protéoglycanes héparane-sulfate hautement sulfatés (HSPG), qui présentent une affinité nanomolaire pour la protéine tau fibrillaire, la dirigeant efficacement vers la voie de dégradation endo-lysosomale (Puangmalai, 2020). Cette capacité est renforcée par la régulation à la hausse du régulateur lysosomal TFEB, qui élargit le compartiment lysosomal des astrocytes, augmentant ainsi leur capacité à traiter la protéine tau englobée (Martini-Stoica, 2018). Il est à noter que l'absorption de la protéine tau par les astrocytes ne se limite pas aux fibrilles. La protéine 1 liée aux lipoprotéines de basse densité (LRP1) agit en coordination avec les HSPG pour internaliser à la fois la protéine tau monomérique et oligomérique, élargissant ainsi la gamme des espèces de protéine tau que les astrocytes peuvent gérer (Reid, 2020). Si ce mécanisme astrocytaire sert initialement à éliminer la protéine tau synaptique et à maintenir l'homéostasie, il crée paradoxalement un réservoir intracellulaire d'agrégats de protéine tau. Ces agrégats capables de se propager peuvent ensuite être libérés, amplifiant la propagation prionique et positionnant les astrocytes comme des acteurs doubles, à la fois protecteurs de l'équilibre synaptique de la protéine tau et contributeurs à sa propagation pathologique.
Les astrocytes de la barrière hémato-encéphalique peuvent également jouer un rôle important dans l'élimination de la protéine tau, notamment grâce à leur connexion au système glymphatique. Leurs pieds terminaux périvasculaires sont enrichis en aquaporine-4 (AQP4), un canal aquifère qui facilite le mouvement du liquide interstitiel et stimule l'échange glymphatique (Sun, 2025). Ce système contribue à éliminer la protéine tau soluble du cerveau, atténuant ainsi la pathologie tau. Cependant, des perturbations de la fonctionnalité de l'AQP4, par le biais de multiples mécanismes, notamment la délétion génétique, la localisation erronée, la fragmentation du sommeil ou le vieillissement, peuvent altérer le flux glymphatique et diminuer l'intégrité de la BHE, réduisant ainsi l'élimination de la protéine tau et exacerbant son accumulation et son agrégation extracellulaires. À l'inverse, les interventions visant à restaurer la polarité de l'AQP4 ou à améliorer la fonction glymphatique se sont révélées prometteuses dans les études précliniques pour réduire la charge tau et améliorer les résultats cognitifs (Zhou, 2025). Par exemple, des approches ciblées, telles que des modifications comportementales, des traitements pharmacologiques et des méthodes neuromodulatrices qui optimisent le pompage glymphatique et l'expression de l'AQP4, fournissent des preuves convaincantes du potentiel thérapeutique (Sun, 2025).
La clairance des agrégats de tau médiée par les cellules gliales peut-elle être améliorée par des moyens thérapeutiques?
Améliorer la clairance des agrégats de protéines tau par les microglies
Les microglies jouent un rôle crucial dans la clairance des protéines tau mal repliées. Des traitements visant à améliorer cette fonction de clairance sont à l'étude. Une piste prometteuse consiste à stimuler le récepteur TREM2, comme le fait l'anticorps monoclonal humain VG-3927, qui fait actuellement l'objet d'études de phase 1 à doses croissantes. En se liant au TREM2, ce traitement vise à restaurer un phénotype microglial homéostatique et à stimuler la clairance phagocytaire de la protéine tau extracellulaire. Une autre approche consiste à inhiber l'inflammasome NLRP3, qui s'est révélée prometteuse dans de nombreux modèles animaux (Zhang, 2020). Quan a montré qu'un déficit en NLPR3 réduit la neuroinflammation et améliore les fonctions cognitives chez des souris transgéniques tau (Quan, 2024)
Parmi les approches les plus avancées figure l'agonisme TREM2: l'anticorps IgG1 entièrement humain VG-3927 est en cours d'achèvement des premières études d'augmentation de la dose et de pharmacodynamique chez l'homme, visant à stimuler les voies de détection des lipides, à restaurer le métabolisme microglial et à améliorer la phagocytose de la protéine tau. Les anticorps précédents (par exemple, AL002 d'Alector et DNL-919 de Denali) ont obtenu des résultats médiocres dans les programmes de phase 2, mais un agoniste TREM2 mis à jour, le VHB937 de Novartis, est entré en phase d'essais pour la MA et la sclérose latérale amyotrophique en 2024, soulignant l'optimisme continu de cette approche. En complément de l'agonisme des récepteurs, l'inhibition de l'inflammasome NLRP3 par la petite molécule MCC950, qui pénètre dans le cerveau, supprime la maturation de l'IL-1β, atténuant ainsi les cascades de chimiokines en aval. Plusieurs inhibiteurs de NLRP3 de nouvelle génération présentant une pharmacocinétique améliorée ont désormais atteint le stade des études permettant l'enregistrement d'un IND.
Pour une analyse approfondie du rôle du TREM2 dans la fonction microgliale, voir: TREM2 et microglies
Approches immunothérapeutiques
Des immunothérapies actives et passives ciblant la protéine tau sont en cours de développement et semblent prometteuses pour moduler la fonction microgliale en remodelant le paysage extracellulaire de la protéine tau (Jadhav, 2019). Les immunothérapies actives, telles que les vaccins peptidiques ou à ARNm, et les immunothérapies passives, notamment les anticorps humanisés, visent à éliminer sélectivement les germes tau solubles. En réduisant les conformères compétents pour la germination ou les fragments tronqués, ces thérapies peuvent influencer indirectement l'activation microgliale, améliorant ainsi l'absorption et l'élimination des agrégats tau sans provoquer de neuroinflammation excessive. Les anticorps de deuxième génération, conçus pour cibler des espèces pathogènes spécifiques de tau, sont en cours d'essais de phase 1 et 2, leur succès dépendant en partie de l'efficacité avec laquelle les microglies traitent la tau opsonisée par les anticorps. Ces stratégies en sont encore à leurs débuts, mais les recherches en cours continuent d'affiner leur potentiel pour le traitement des maladies neurodégénératives associées à la tau.
Cibles astrocytaires: du flux glymphatique à la protéostasie
Les astrocytes facilitent l'élimination en masse des solutés interstitiels via un échange périvasculaire dépendant de l'AQP4 et sécrètent des protéines de choc thermique qui chaperonnent la protéine tau mal repliée (Zhou, 2025). La régulation à la hausse expérimentale de la polarité de l'AQP4 ou l'activation pharmacologique du TFEB astrocytaire (un régulateur maître de la biogenèse lysosomale) accélère l'élimination de la protéine tau dans des modèles murins de tauopathie et est actuellement explorée à l'aide d'agonistes TFEB à petites molécules et de modulateurs de l'AQP4 (Verkman, 2013). En outre, la conversion astrocytaire des cytokines libérées par les microglies (par exemple, les programmes de clairance des lipides induits par l'IL-33) met en évidence l'interaction bidirectionnelle entre les cellules gliales comme approche thérapeutique potentielle (Carlock, 2017; Sun, 2021).
Modulation métabolique du traitement post-traductionnel de la protéine tau
Au-delà de l'inflammation, la modulation métabolique de la protéine tau elle-même reste une cible thérapeutique intéressante . L'inhibition sélective de l'O-GlcNAcase (OGA) augmente l'O-GlcNAcylation de la protéine tau, empêchant ainsi stériquement la phosphorylation et l'agrégation pathologiques (Kielbasa, 2024; Selnick, 2019; Wang, 2020). Le MK-8719, un inhibiteur de l'OGA puissant et sélectif à petite molécule disponible par voie orale, est passé à la phase 1b des essais cliniques dans le traitement de la PSP ; bien que les premiers signes d'efficacité aient été modestes, les biomarqueurs pharmacodynamiques du liquide céphalo-rachidien (LCR) ont confirmé une forte interaction avec la cible. Un deuxième inhibiteur de l'OGA, le ceperognastat (anciennement MK-8722), a été interrompu par Eli Lilly en février 2025 après une exposition sous-thérapeutique dans une cohorte de phase 2 atteinte de la MA, mais des améliorations de la structure et de l'activité sont en cours. Il est important de noter que ces interventions métaboliques reposent sur le mécanisme fonctionnel de la protéostasie gliale, ce qui renforce le concept selon lequel le soutien des microglies et des astrocytes est une condition préalable à un bénéfice thérapeutique maximal.
Perspectives
Collectivement, les données issues de l'agonisme des récepteurs, du blocage de l'inflammasome, de l'immunothérapie et de la modulation métabolique convergent vers un thème central : les cellules gliales possèdent une capacité sous-exploitée à neutraliser la protéine tau pathogène, mais cette capacité doit être réorientée vers un phénotype équilibré et homéostatique. Les essais futurs devront combiner un engagement moléculaire précis avec des biomarqueurs fluides et d'imagerie sensibles afin de capturer les changements en temps réel de l'état des cellules gliales et de la charge tau. La question de savoir si les modulateurs gliaux à agent unique peuvent ralentir de manière cliniquement significative la progression de la maladie ou s'ils seront plus efficaces en association rationnelle avec des anticorps dirigés contre la protéine tau ou des oligonucléotides antisens reste ouverte. Il est important de noter que ce domaine évolue rapidement et que d'autres candidats thérapeutiques ciblant à la fois les interactions entre les astrocytes et la protéine tau dans la microglie sont à l'étude. Les recherches en cours continuent de mettre en lumière le rôle complexe des cellules gliales dans les tauopathies, dans le but de développer des traitements efficaces modifiant le cours de la maladie.
Notre équipe se fera un plaisir de répondre à toutes vos questions concernant les microglies, les astrocytes et la protéine tau dans les maladies neurodégénératives, ou de vous fournir des informations spécifiques sur les modèles de MA et de tauopathies que nous utilisons pour nos études d'efficacité thérapeutique.
En savoir plus sur nos modèles de maladies neurodégénératives
Contenu connexe
Informations actualisées sur la neuroinflammation et les meilleures pratiques liées à l'évaluation des agents thérapeutiques dans des modèles animaux de maladies neurodégénératives.
TREM2 et microglies
Présentation générale du TREM2, de son rôle dans les microglies, de ses liens avec les maladies neurodégénératives et de ses implications potentielles en matière de traitement.
Interactions entre les microglies et les neurones et maladies neurodégénératives
Une revue concise des interactions directes entre les microglies et les neurones, et de la manière dont ces interactions intercellulaires peuvent être affectées dans les maladies neurodégénératives.
Microglie, astrocytes et α-synucléine dans la maladie de Parkinson
Comment l'α-synucléine influence les microglies et les astrocytes dans la maladie de Parkinson et d'autres synucléinopathies.
TNF-α et microglie dans les maladies neurodégénératives
Un aperçu de la fonction du facteur de nécrose tumorale alpha (TNF-α) dans la microglie et de sa contribution à la progression de la neurodégénérescence.
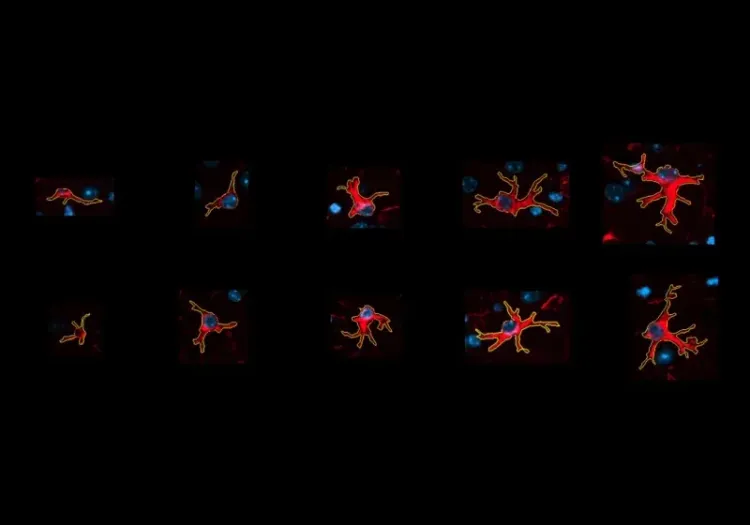
Astrocytes et modèles murins β amyloïde de la maladie d’Alzheimer
L'analyse de la morphologie des astrocytes dans le microenvironnement de la plaque amyloïde-β fournit une mesure sensible de la progression de la maladie chez les souris transgéniques.