Dysfonctionnement mitochondrial dans les microglies et les astrocytes
Le rôle du dysfonctionnement mitochondrial dans les microglies et les astrocytes dans les maladies neurodégénératives, notamment la maladie d'Alzheimer, la maladie de Parkinson et la SLA.
Cette ressource décrit :
- Quel est le rôle des mitochondries dans les microglies et les astrocytes?
- Quel est le lien entre le dysfonctionnement mitochondrial dans les microglies et les astrocytes et la MA, la MP et la SLA?
- Quelles sont les approches thérapeutiques ciblant le dysfonctionnement mitochondrial dans les maladies neurodégénératives?
Quel est le rôle des mitochondries dans les microglies et les astrocytes?
Fonction et dynamique mitochondriales
Les mitochondries sont des organites essentiels qui soutiennent un large éventail de fonctions cellulaires. Surnommées les « centrales énergétiques » de la cellule, les mitochondries produisent de l'adénosine 5'-triphosphate (ATP), la principale source d'énergie des processus cellulaires, principalement par le biais de la phosphorylation oxydative (OXPHOS) via la chaîne de transport d'électrons (ETC), qui comprend une série de complexes protéiques (Tönnies, 2017). Cependant, leurs rôles vont bien au-delà de la production d'énergie. Les mitochondries sont également impliquées dans le métabolisme des lipides et des acides aminés, l'homéostasie du calcium (Ca2+) et la mort cellulaire programmée (PCD), souvent appelée apoptose (Bose, 2016). De plus, elles modulent le stress oxydatif en équilibrant la production d'espèces réactives de l'oxygène (ERO) et les défenses antioxydantes.
Les mitochondries sont très dynamiques et subissent un remodelage morphologique continu par des processus de fusion et de fission. Ces changements dynamiques sont essentiels au maintien de la fonction mitochondriale et à la prévention de l'apoptose. Les protéines clés impliquées dans la dynamique mitochondriale comprennent la protéine 1 apparentée à la dynamine (Drp1) pour la fission, et les mitofusines (Mfn1/2) et l'atrophie optique 1 (OPA1) pour la fusion. Une perturbation de l'équilibre de ces processus peut entraîner une fragmentation mitochondriale, une réduction de la production d'ATP et une augmentation du stress cellulaire. En outre, une fission et une fusion défectueuses peuvent altérer le transport mitochondrial, contribuant ainsi à un dysfonctionnement synaptique et à une neurodégénérescence.
Dysfonctionnement mitochondrial et neuroinflammation à médiation gliale
Le dysfonctionnement mitochondrial peut résulter de divers facteurs, notamment le vieillissement et des mutations de l'ADN mitochondrial (ADNmt), entraînant une altération de la production d'ATP, une augmentation des niveaux de ROS, des anomalies structurelles, un stress oxydatif et l'apoptose (Tönnies, 2017; Wang, 2020). Si les niveaux physiologiques de ROS servent de molécules de signalisation, un excès de ROS perturbe l'intégrité et le fonctionnement des mitochondries (Kumar, 2012). Les mitochondries endommagées qui ne sont pas éliminées de manière adéquate par la mitophagie peuvent libérer des motifs moléculaires associés aux dommages (DAMP) dérivés des mitochondries, qui déclenchent des voies de signalisation inflammatoire par l'activation de récepteurs immunitaires innés (Salmina, 2021; Lin, 2022).
Les microglies et les astrocytes jouent un rôle essentiel dans la réponse immunitaire du système nerveux central (SNC) et sont les principaux répondeurs aux DAMPs mitochondriaux. Les microglies reconnaissent ces signaux de danger grâce à des récepteurs de reconnaissance de motifs, activant des facteurs de transcription tels que NF-κB, qui déclenchent la libération de cytokines pro-inflammatoires comme l'interleukine-1β (IL-1β) et le facteur de nécrose tumorale alpha (TNF-α). Ces cytokines exacerbent le stress oxydatif, perturbent la barrière hémato-encéphalique (BHE) et favorisent les lésions neuronales (Wang, 2020). Dans les microglies comme dans les astrocytes, le dysfonctionnement mitochondrial altère les fonctions neuroprotectrices et entretient une inflammation chronique. Il en résulte un cercle vicieux dans lequel les dommages mitochondriaux activent les cellules gliales, et les médiateurs inflammatoires qui en résultent endommagent davantage les mitochondries, amplifiant ainsi la neurodégénérescence (Jeon, 2020; Yan, 2020).
Il est important de noter que le dysfonctionnement mitochondrial peut précéder la neuroinflammation dans les maladies neurodégénératives telles que la maladie d'Alzheimer (MA), la maladie de Parkinson (MP) et la sclérose latérale amyotrophique (SLA), ce qui positionne le dysfonctionnement mitochondrial comme un facteur potentiel de la pathologie de ces maladies (Swerdlow, 2014). L'étude des changements mitochondriaux dans les cellules gliales, en plus des neurones, pourrait permettre de mieux comprendre les mécanismes précoces de la maladie et faciliter le développement de nouvelles stratégies thérapeutiques (Lin, 2022).
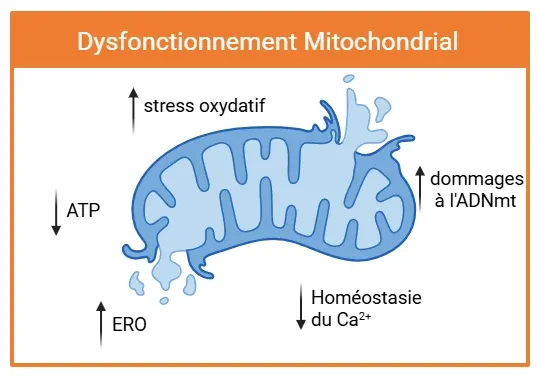
La dysfonction mitochondriale se caractérise par des lésions structurelles et un dysfonctionnement. Les flèches indiquent les principaux changements pathologiques, notamment l'augmentation de la production de ERO, le stress oxydatif et les dommages de l'ADNmt. Ces altérations coïncident avec une diminution de la production d'ATP et une perturbation de l'homéostasie du Ca2+, contribuant à la neuroinflammation et à la progression des maladies neurodégénératives.
Quel est le lien entre le dysfonctionnement mitochondrial dans les microglies et les astrocytes et la MA, la MP et la SLA?
Le dysfonctionnement mitochondrial dans les microglies et les astrocytes est un domaine d'intérêt émergent dans l'étude des maladies neurodégénératives. Une altération de la fonction mitochondriale dans ces cellules gliales nuit non seulement à leur rôle de soutien, mais déclenche et entretient également une neuroinflammation, créant ainsi une boucle de rétroaction qui aggrave les lésions neuronales.
Maladie d'Alzheimer (MA)
La MA se caractérise cliniquement par une perte progressive de la mémoire, un déclin cognitif et des changements comportementaux. Ses signes pathologiques comprennent l'accumulation de plaques amyloïdes bêta (Aβ), des enchevêtrements neurofibrillaires hyperphosphorylés de protéine tau et la perte neuronale. Il convient de noter que le dysfonctionnement mitochondrial, le stress oxydatif et l'inflammation comptent parmi les premières caractéristiques contribuant à la progression de la MA (Swerdlow, 2014).
Dans la MA, l'accumulation d'Aβ dans les mitochondries inhibe les complexes II et IV de la chaîne de transport d'électrons, réduisant la production d'ATP et augmentant les ROS (Kumar, 2015; Swerdlow, 2016). L'Aβ perturbe également l'importation des protéines mitochondriales, endommage l'ADNmt et altère l'homéostasie du Ca2+, favorisant ainsi l'apoptose (Kumar, 2015). En outre, l'Aβ perturbe la morphologie et la dynamique mitochondriales, y compris la régulation de la fission et de la fusion (Wang, 2008; 2009). La dérégulation de Drp1 et OPA1, qui interviennent dans ces dynamiques, est liée à la toxicité neuronale induite par l'Aβ (Wang, 2008; 2009). L'altération de l'élimination des mitochondries endommagées amplifie la neurodégénérescence (Kumar, 2015).
Les modèles précliniques suggèrent que les interventions ciblant les mitochondries pourraient offrir un bénéfice thérapeutique. Par exemple, la surexpression de l'OPA1 restaure la structure et la fonction mitochondriales dans les modèles de MA (Wang, 2008; 2020). De plus, le P110, un inhibiteur sélectif de Drp1/Fis1, réduit la fragmentation mitochondriale et l'activation microgliale et astrocytaire, ce qui diminue la libération de mitochondries endommagées par les microglies et protège les neurones contre les lésions médiées par les cytokines (Joshi, 2019). Ces résultats confirment le potentiel de cibler la fission mitochondriale dans les microglies pour atténuer la neuroinflammation et les lésions neuronales.
Maladie de Parkinson (MP)
La MP se manifeste par des symptômes moteurs tels que la bradykinésie, la rigidité, des difficultés à marcher et des tremblements au repos, ainsi que des symptômes non moteurs tels que des troubles cognitifs et des troubles de l'humeur et du sommeil. Elle se caractérise par l'accumulation d'alpha-synucléine (alpha-syn) dans les corps de Lewy et la dégénérescence des neurones dopaminergiques dans la substance noire compacte (SNpc). Si la plupart des cas de MP sont sporadiques, certains sont associés à des mutations génétiques dans des gènes tels que DJ-1 (également connu sous le nom de PARK7) (Almikhalfi, 2020).
La MP familiale et sporadique présente des anomalies mitochondriales (Grünewald, 2019). Les rats knock-out DJ-1 présentent une neurodégénérescence dopaminergique progressive dans la SN, des déficits moteurs et des perturbations des voies mitochondriales, telles que le complexe I de la chaîne de transport d'électrons (Almikhlafi, 2020). Les astrocytes contribuent à la dégradation de l'alpha-synéphrine par la voie autophagie-lysosome; cependant, un excès d'alpha-synucléine perturbe ce processus, entraînant une dysfonction mitochondriale supplémentaire. L'alpha-synucléine interagit avec des composants mitochondriaux, notamment la cytochrome C oxydase 1 (COX1), altérant ainsi la fonction mitochondriale (Jeon, 2020). Les mutations du gène ATP13A2 perturbent également les voies mitochondriales et lysosomales, contribuant à l'accumulation d'alpha-syn (Grünewald, 2019).
Sclérose latérale amyotrophique (SLA)
La SLA est une maladie neurodégénérative progressive qui affecte les motoneurones, entraînant une faiblesse musculaire, une spasticité et une insuffisance respiratoire. Si la plupart des cas sont sporadiques, la SLA familiale est liée à des mutations dans des gènes tels que C9orf72, SOD1, FUS et TARDBP. Les protéines mal repliées, comme la superoxyde dismutase 1 (SOD1) mutante et la TDP-43, sont des caractéristiques marquantes de la SLA.
Une morphologie mitochondriale anormale a été observée chez des patients atteints de SLA, chez des souris et dans des modèles de culture cellulaire (Magrané, 2009). Le transport et le positionnement des mitochondries au niveau des synapses de la jonction neuromusculaire sont essentiels au fonctionnement des motoneurones. Des études post mortem montrent une activité réduite du complexe ETC dans la moelle épinière des patients atteints de SLA (Obrador, 2020). La dérégulation des sirtuines mitochondriales, telles que SIRT1 et SIRT3, altère la biogenèse et le fonctionnement des mitochondries (Obrador, 2020). SIRT1 active PGC-1α, qui améliore le fonctionnement des mitochondries et la défense contre les ROS via SIRT3 (Song, 2013). L'activation de SIRT1 par le resvératrol améliore les symptômes et la survie dans le modèle murin de SLA SOD1G93A, tandis que la surexpression de SIRT3 diminue la production de ROS et protège contre la fragmentation et la mort mitochondriales dans les motoneurones SOD1G93A en culture (Han, 2012; Song, 2013).
La TDP-43 interagit également avec des protéines mitochondriales, telles que le canal anionique voltage-dépendant 1 (VDAC1), perturbant la dynamique mitochondriale et la mitophagie (Obrador, 2020). Dans les motoneurones C9orf72 dérivés de cellules souches pluripotentes induites (iPSC), le stress oxydatif augmente avec l'âge, mais peut être partiellement atténué en réduisant le stress oxydatif (Lopez-Gonzalez, 2016).
En résumé, bien que la MA, la MP et la SLA diffèrent dans leur présentation et leur pathologie, elles partagent un mécanisme commun: un dysfonctionnement mitochondrial qui entraîne une activation gliale chronique et une neuroinflammation. Cette relation bidirectionnelle crée une boucle de rétroaction qui accélère la progression de la maladie. Cibler ce cycle pourrait offrir de nouvelles pistes thérapeutiques.
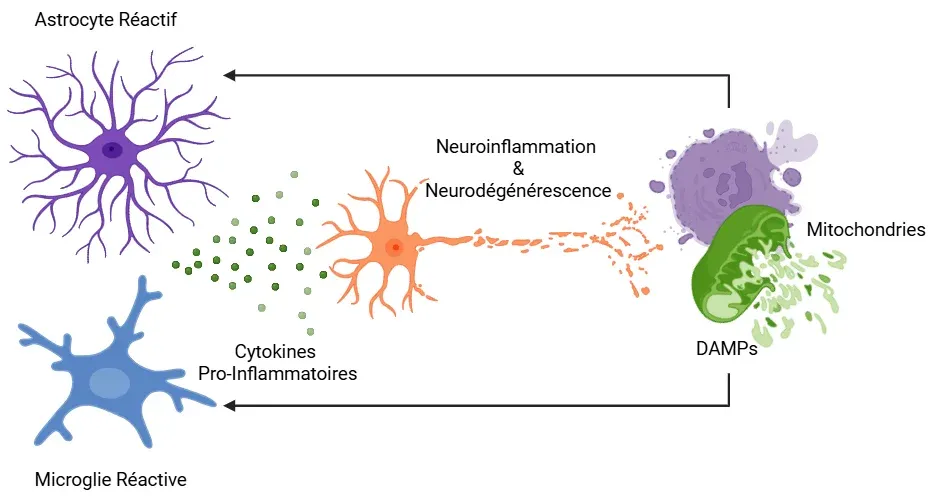
La neuroinflammation dans les maladies neurodégénératives est provoquée par l'interaction entre des mitochondries dysfonctionnelles et des cellules gliales réactives. Les mitochondries endommagées libèrent des DAMPs dérivés des mitochondries, qui sont reconnus par les microglies et les astrocytes. En réponse, ces cellules gliales adoptent un phénotype réactif et sécrètent des cytokines pro-inflammatoires, ce qui altère davantage la fonction mitochondriale. Ce processus établit une boucle de rétroaction auto-entretenue dans laquelle le dysfonctionnement mitochondrial et la réactivité gliale s'amplifient mutuellement, contribuant à une neuroinflammation chronique et à une dégénérescence neuronale progressive. Figure adaptée de Picca et al. (Picca, 2022) sous licence Creative Commons Attribution.
Quelles sont les approches thérapeutiques ciblant le dysfonctionnement mitochondrial dans les maladies neurodégénératives?
Compte tenu du rôle central du dysfonctionnement mitochondrial et du stress oxydatif dans les maladies neurodégénératives, le ciblage des voies mitochondriales est apparu comme une stratégie thérapeutique prometteuse. Plusieurs approches, telles que les antioxydants ciblant les mitochondries, la modulation de la dynamique mitochondriale et l'amélioration de la fonction bioénergétique, sont actuellement à l'étude. Bien que des défis tels que la pénétration de la BHE persistent, ces interventions offrent un potentiel pour ralentir ou modifier la progression de la maladie.
Le stress oxydatif, résultant d'un déséquilibre entre les ROS et les défenses antioxydantes, est un facteur clé du dysfonctionnement mitochondrial. Les antioxydants ciblant les mitochondries ont donné des résultats encourageants dans des modèles précliniques en prévenant les dommages oxydatifs aux neurones (Kumar, 2015). Des composés tels que MitoQ, MitoTEMPOL et mitovitE sont conçus pour atténuer le stress oxydatif à la source, au sein des mitochondries. Cependant, leur capacité limitée à traverser la BHE reste un obstacle important (Hara, 2019). De plus, la thérapie antioxydante doit être soigneusement régulée, car une activité antioxydante excessive peut perturber les voies de signalisation oxydatives essentielles au fonctionnement normal des cellules (Hara, 2019).
Les processus dynamiques de fission et de fusion mitochondriales sont essentiels au maintien de la santé mitochondriale. La dérégulation de ces processus a été impliquée dans plusieurs maladies neurodégénératives. Le ciblage thérapeutique des protéines impliquées dans la dynamique mitochondriale, telles que Drp1 ou OPA1, a gagné en popularité. Par exemple, l'inhibition de Drp1 par la petite molécule Mdivi-1 a montré qu'elle restaurait la morphologie mitochondriale, améliorait la dynamique, réduisait la pathologie amyloïde et améliorait les performances cognitives chez des modèles murins de MA à un stade précoce, probablement en réduisant la production de Aβ (Wang, 2020).
Une autre stratégie thérapeutique émergente se concentre sur la modulation de la bioénergétique mitochondriale. L'amélioration de la fonction mitochondriale et de la production d'ATP pourrait aider à compenser les déficits énergétiques couramment observés dans les maladies neurodégénératives. L'oxaloacétate (OAA), un intermédiaire du cycle de Krebs, a démontré sa capacité à augmenter le flux bioénergétique mitochondrial et à améliorer la fonction cérébrale. Bien qu'un essai clinique de phase I chez des patients atteints de MA ait confirmé son innocuité, ses bénéfices cognitifs étaient limités (Swerdlow, 2016). Des études supplémentaires sont nécessaires pour déterminer la posologie efficace et l'efficacité à long terme. Un autre candidat, le CP2, un composé qui s'accumule sélectivement dans les mitochondries neuronales et inhibe le complexe I mitochondrial, a montré un potentiel dans des modèles murins de MA en réduisant les plaques amyloïdes et la pathologie tau et en préservant la fonction cognitive (Zhang, 2015).
Les thérapies à base de peptides, telles que le SS-31 (également connu sous le nom d'elamipretide ou MTP-131), représentent une autre approche innovante. Le SS-31 cible la membrane mitochondriale interne et se lie à la cardiolipine, un lipide essentiel à l'intégrité de la membrane mitochondriale. Cette liaison favorise la formation de supercomplexes impliqués dans l'OXPHOS. Des études précliniques ont montré que le SS-31 améliore la fonction mitochondriale et synaptique dans des modèles de MA, et il a récemment été testé dans des essais cliniques pour des maladies rares, telles que l'ataxie de Friedreich (NCT05168774).
En conclusion, les stratégies thérapeutiques visant à corriger le dysfonctionnement mitochondrial sont très prometteuses pour le traitement des maladies neurodégénératives. Cependant, plusieurs défis restent à relever, notamment l'optimisation de l'administration des médicaments, la garantie de la pénétration de la BHE et le maintien d'un équilibre entre la suppression des ROS et la signalisation physiologique. Il est essentiel de poursuivre les recherches afin d'affiner ces thérapies et de les traduire en traitements efficaces pour des maladies telles que la MA, la MP et la SLA.
Notre équipe se fera un plaisir de répondre à toutes vos questions concernant le dysfonctionnement mitochondrial dans les microglies et les astrocytes et les maladies neurodégénératives, ou de vous fournir des informations spécifiques sur les modèles de MA, SLA et MP que nous utilisons pour nos études d'efficacité thérapeutique.
En savoir plus sur nos modèles de maladies neurodégénératives
Contenu connexe
Informations actualisées sur le dysfonctionnement mitochondrial dans les microglies et les astrocytes et les maladies neurodégénératives, ainsi que les meilleures pratiques en matière d'évaluation des agents thérapeutiques dans les modèles animaux de maladies neurodégénératives.
Interactions entre les microglies et les neurones et maladies neurodégénératives
Une revue concise des interactions directes entre les microglies et les neurones, et de la manière dont ces interactions intercellulaires peuvent être affectées dans les maladies neurodégénératives.
Microglie, astrocytes et α-synucléine dans la maladie de Parkinson
Comment l'α-synucléine influence les microglies et les astrocytes dans la maladie de Parkinson et d'autres synucléinopathies.
Dysfonctionnement lysosomal dans les microglies et les astrocytes
Un aperçu du dysfonctionnement lysosomal dans la microglie et les astrocytes, et de son rôle dans les maladies neurodégénératives.
TNF-α et microglie dans les maladies neurodégénératives
Un aperçu de la fonction du facteur de nécrose tumorale alpha (TNF-α) dans la microglie et de sa contribution à la progression de la neurodégénérescence.
Astrocytes et modèles murins β amyloïde de la maladie d’Alzheimer
L'analyse de la morphologie des astrocytes dans le microenvironnement de la plaque amyloïde-β fournit une mesure sensible de la progression de la maladie chez les souris transgéniques.
Dysfonctionnement mitochondrial et maladie de Parkinson
Un aperçu de la façon dont le dysfonctionnement mitochondrial est associé à la neurodégénérescence dans la maladie de Parkinson.
Mitophagie et maladie de Parkinson
Une vue d'ensemble de la façon dont une mitophagie déficiente peut conduire à la neurodégénérescence dans la maladie de Parkinson.