TNF-α et microglie dans les maladies neurodégénératives
Un aperçu de la fonction du facteur de nécrose tumorale alpha (TNF-α) dans la microglie et de sa contribution à la progression de la neurodégénérescence.
Cette ressource décrit:
Quel est le rôle du TNF-α dans le système nerveux central (SNC)?
Le facteur de nécrose tumorale alpha (TNF-α) est une cytokine pro-inflammatoire qui joue un rôle crucial dans la régulation de l'inflammation et des réponses immunitaires. Dans le système nerveux central (SNC), le TNF-α a une double fonction, contribuant positivement dans des conditions saines, mais pouvant causer des dommages lorsqu'il est dérégulé dans des états pathologiques. Dans des conditions homéostatiques, le TNF-α favorise la neuroplasticité, la myélinisation et la réparation des tissus. Cependant, lorsque ses niveaux deviennent élevés, il peut entraîner des effets néfastes, tels que l'excitotoxicité, l'inflammation chronique et la rupture de la barrière hémato-encéphalique (BHE). Le TNF-α existe sous forme soluble et transmembranaire, et agit par l'intermédiaire de deux récepteurs principaux : Le récepteur TNF de type 1 (TNFR1) et le récepteur TNF de type 2 (TNFR2). Son expression est étroitement régulée par divers facteurs de transcription, notamment le facteur nucléaire kappa-light-chain-enhancer des cellules B activées (NF-κB). Lorsqu'il est activé, le NF-κB favorise non seulement la production de TNF-α, mais déclenche également l'expression d'autres cytokines pro-inflammatoires, telles que l'IL-6 et l'IL-1β. Cette interaction entre le TNF-α et le NF-κB crée une boucle de rétroaction positive, qui amplifie la réponse inflammatoire et soutient la réaction immunitaire (Gonzalez Caldito, 2023).
Les microglies, les cellules immunitaires résidentes du SNC, sont la principale source de TNF-α pendant la neuroinflammation, bien que les astrocytes puissent également contribuer à sa libération (Olmos, 2014). Dans les maladies neurodégénératives, la réactivité microgliale, ainsi que d'autres états tels que la sénescence microgliale et le phénotype sécrétoire associé à la sénescence (SASP), entraînent une production accrue de TNF-α et d'autres cytokines pro-inflammatoires, entretenant un environnement inflammatoire chronique. Les cytokines inflammatoires, telles que l'interféron-gamma (IFN-&up;), peuvent en outre stimuler la microglie à libérer du TNF-α, exacerbant ainsi l'inflammation. Cette réponse inflammatoire continue accélère la progression des maladies neurodégénératives, telles que la maladie d'Alzheimer (MA), la maladie de Parkinson (MP) et la sclérose latérale amyotrophique (SLA).
Les stratégies thérapeutiques ciblant le TNF-α ont montré leur potentiel dans le traitement des maladies auto-immunes du SNC et pourraient permettre de modifier les caractéristiques neuropathologiques des maladies neurodégénératives (Torres-Acosta, 2020; Gonzalez Caldito, 2023). Cependant, si les traitements anti-TNF-α peuvent réduire l'inflammation, leurs effets sont complexes et peuvent parfois entraîner des conséquences inattendues, comme l'exacerbation de maladies telles que la sclérose en plaques (SEP) (Fresegna, 2020). Ces effets secondaires sont potentiellement dus au double rôle du TNF-α et de ses récepteurs, qui peuvent avoir à la fois des effets pro-inflammatoires et protecteurs selon le contexte. En outre, les recherches sur la régulation temporelle du TNF-α, en particulier aux stades précoces et tardifs de la progression de la maladie, pourraient permettre de comprendre comment le moment de la modulation du TNF-α peut influer sur les résultats du traitement (Guidotti, 2021). Par conséquent, une meilleure compréhension du rôle du TNF-α dans le SNC et des recherches plus approfondies sur la sécurité et l'efficacité de ces thérapies sont nécessaires pour améliorer les stratégies de traitement.
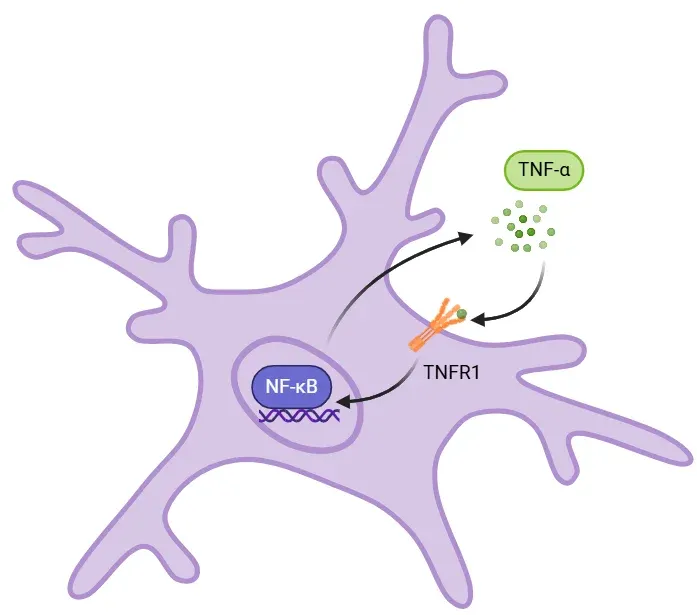
Lorsque le TNF-α soluble se lie au TNFR1, il déclenche une cascade de signaux (non représentée) qui active le NF-κB. Dans le noyau, le NF-κB entraîne la transcription de cytokines pro-inflammatoires, notamment l'IL-6, l'IL-1β et le TNF-α lui-même. Cette régulation positive de la cytokine amplifie la réponse immunitaire par une boucle de rétroaction positive où le TNF-α augmente l'activité du NF-κB, ce qui, à son tour, favorise la production de TNF-α. Des taux élevés de TNF-α peuvent entraîner des lésions tissulaires, une excitotoxicité et d'autres effets nocifs. Lorsqu'elle est dérégulée, comme dans les maladies neurodégénératives, cette boucle entretient une inflammation chronique.
Comment le TNF-α est-il impliqué dans les maladies neurodégénératives telles que la MA, la MP et la SLA ?
La maladie d'Alzheimer (MA)
La maladie d'Alzheimer (MA) est une maladie neurodégénérative caractérisée par un déclin cognitif progressif, des troubles de la mémoire et l'accumulation de plaques amyloïdes bêta (Aβ) et d'enchevêtrements neurofibrillaires tau. De plus en plus de preuves suggèrent que les cytokines inflammatoires jouent un rôle central dans la physiopathologie de la MA. Des taux élevés de TNF-α, d'IL-1β et d'IL-6 ont été détectés dans le sang et le liquide céphalorachidien (LCR) de patients atteints de MA (Brosseron, 2014). Cependant, certaines études ont rapporté des résultats contradictoires, potentiellement dus à des différences de stade de la maladie. Malgré ces divergences, les cytokines telles que le TNF-α ont tendance à augmenter à mesure que la MA progresse, bien que leurs taux soient souvent trop faibles pour servir de biomarqueurs fiables. La relation précise entre les taux de cytokines et la progression de la maladie fait toujours l'objet de recherches. Certaines études suggèrent que des taux élevés de cytokines peuvent indiquer un risque accru de développer la MA, et que les ratios des cytokines par rapport à leurs récepteurs respectifs peuvent fournir des marqueurs diagnostiques plus précis que les taux individuels de cytokines seuls (Brosseron, 2014).
Le dysfonctionnement microglial est un autre facteur critique dans la pathogenèse de la MA. Dans les modèles d'accumulation d'Aβ, la microglie âgée présente une activité phagocytaire altérée et une expression accrue des cytokines inflammatoires, notamment le TNF-α et l'IL-1β (Hickman, 2008). Ce dysfonctionnement déclenche une boucle de rétroaction dans laquelle l'accumulation de plaques Aβ active la microglie, l'amenant à libérer du TNF-α. Cette libération de cytokines amplifie la neuroinflammation et accélère les dommages neuronaux. La libération soutenue de cytokines favorise la poursuite de la neurodégénérescence, ce qui, à son tour, aggrave la réponse inflammatoire, contribuant à l'augmentation des dépôts d'Aβ. L'incapacité de la microglie à éliminer efficacement les plaques d'Aβ entraîne l'accumulation d'agrégats toxiques. Ce cycle d'accumulation de plaques, de réactivité microgliale, de libération de TNF-α et de dommages neuronaux perpétue le processus neurodégénératif, accélérant la progression de la MA (Hickman, 2008).
Les essais cliniques étudiant le potentiel thérapeutique de l'inhibition du TNF-α dans la MA ont donné des résultats mitigés. De petites études ouvertes ont montré des résultats prometteurs lorsque l'inhibiteur du TNF-α étanercept a été administré par voie périspinale, montrant des améliorations de la cognition et du comportement (Tobinick, 2008). Cependant, lorsque le même traitement a été administré par voie périphérique, aucun changement significatif n'a été observé dans ces domaines (Butchart, 2015). Cependant, un essai de phase 1b sur le XPro1595, un inhibiteur du TNF-α qui cible sélectivement le TNF-α soluble, a montré des résultats prometteurs dans la MA (NCT03943264). Cette nouvelle approche, qui se concentre sur la forme soluble du TNF-α, pourrait offrir une méthode plus ciblée et plus efficace pour moduler la réponse inflammatoire associée à la MA.
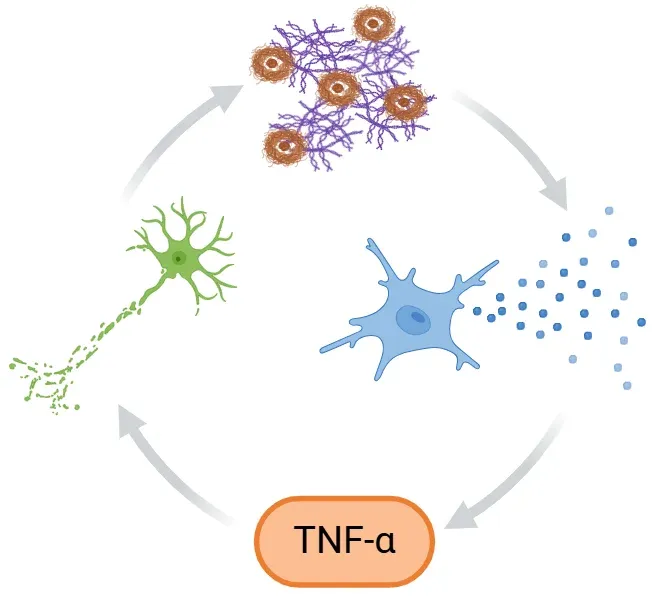
Boucle de rétroaction positive proposée dans la maladie d'Alzheimer (MA), où les plaques Aβ et les enchevêtrements tau activent la microglie, libérant des cytokines pro-inflammatoires comme le TNF-α. Cette libération amplifie la neuroinflammation et la neurodégénérescence, tandis que l'altération de la clairance microgliale augmente l'accumulation de protéines toxiques, aggravant encore le cycle et accélérant la progression de la MA.
La maladie de Parkinson (MP)
La maladie de Parkinson est une maladie neurodégénérative progressive caractérisée par la perte de neurones dopaminergiques dans la substance noire compacte (SNpc), entraînant des symptômes moteurs tels que des tremblements, une rigidité et une bradykinésie. Outre ces symptômes moteurs caractéristiques, la maladie de Parkinson est également associée à une série de symptômes non moteurs, notamment un déclin cognitif, des troubles du sommeil et une dépression.
La neuroinflammation chronique a été identifiée comme un facteur clé dans la physiopathologie de la MP. L'analyse post-mortem du SN de patients atteints de MP révèle une réactivité microgliale et des niveaux élevés de cytokines pro-inflammatoires, telles que le TNF-α, qui sont également présents dans le sérum des patients atteints de MP (Collins, 2012). Le TNF-α est exprimé sur les neurones dopaminergiques dans le SNpc humain, ce qui suggère en outre que le TNF-α joue un rôle dans le processus pathologique. En outre, les modèles animaux de la MP montrent une augmentation de l'expression du TNF-α à la fois dans le sérum et le LCR (Collins, 2012).
L'activation du facteur de transcription NF-κB, qui régule les gènes codant pour les cytokines pro-inflammatoires, est également élevée chez les patients atteints de la MP, ce qui relie davantage les voies inflammatoires à cette maladie (Mogi, 2007). Des recherches ont montré que la microglie réactive chronique libère des niveaux élevés de cytokines pro-inflammatoires telles que le TNF-α, qui non seulement endommagent les neurones dopaminergiques, mais perpétuent également une boucle de rétroaction qui entraîne une réactivité microgliale, une neuroinflammation et une neurodégénérescence supplémentaires dans la MP (Collins, 2012).
Des études sur des modèles animaux ont démontré que le blocage du TNF-α peut atténuer considérablement la neurodégénérescence dans la MP. Par exemple, le traitement à la thalidomide, un inhibiteur de la synthèse du TNF-α, protège contre la déplétion dopaminergique induite par le MPTP, et les souris déficientes en TNF-α présentent une réduction significative de la perte de dopamine striatale et de la mortalité (Ferger, 2004). De plus, il a été démontré que l'utilisation de XENP345, un inhibiteur du TNF-α, réduit la mort des neurones dopaminergiques dans les modèles murins de la MP (McCoy, 2006). Ces résultats suggèrent que le ciblage du TNF-α pourrait être une approche thérapeutique prometteuse pour moduler les processus inflammatoires et ralentir la progression de la maladie dans la MP.
Sclérose latérale amyotrophique (SLA)
La SLA est une maladie neurodégénérative progressive qui cible principalement les motoneurones, entraînant une faiblesse musculaire, des difficultés à parler et à avaler et, finalement, une spasticité et une paralysie. En plus de ces caractéristiques principales, la neuroinflammation joue un rôle important dans la progression de la maladie. Des taux élevés de TNF-α ont été détectés dans le plasma et le sérum de patients atteints de SLA, ainsi que dans des modèles animaux de la maladie (Olmos, 2014; Vu, 2017). De plus, la régulation positive de NF-κB a été observée dans la microglie de la moelle épinière de patients atteints de SLA et dans le modèle de souris transgénique superoxyde dismutase 1 (SOD1), un modèle couramment utilisé pour étudier la SLA (Frakes, 2014). Ces résultats confirment l'implication du TNF-α dans la physiopathologie de la SLA.
Pour mieux comprendre le rôle du TNF-α dans la SLA, des études de suppression de gènes ont été menées, en particulier sur le modèle de souris transgénique SOD1. Ces études n'ont révélé aucun impact significatif sur la durée de vie ou la dégénérescence des motoneurones à la suite de la suppression génétique du TNF-α (Gowing, 2006). Ces résultats suggèrent que, si le TNF-α peut contribuer aux processus inflammatoires observés dans la SLA, il pourrait ne pas être directement responsable de la dégénérescence des motoneurones, en particulier dans le contexte des mutations SOD1.
D'autre part, des recherches utilisant des inhibiteurs du TNF-α, comme la thalidomide et la lénalidomide, ont donné des résultats prometteurs. Chez les souris transgéniques SOD1, ces inhibiteurs ont amélioré les performances motrices et diminué la mort des cellules motrices (Kiaei, 2006). Ces résultats suggèrent que le ciblage du TNF-α pourrait être une approche thérapeutique viable pour la SLA. Cependant, un essai clinique de phase 2 portant sur l'utilisation de la thalidomide chez des patients atteints de SLA n'a pas reproduit ces résultats prometteurs. L'administration quotidienne de thalidomide n'a pas montré d'effets bénéfiques et a été associée à plusieurs effets secondaires indésirables (Stommel, 2009).
Ces résultats mitigés peuvent être dus au moment du traitement, car l'efficacité de l'inhibition du TNF-α peut varier en fonction du stade de progression de la SLA, ce qui souligne la nécessité de poursuivre les recherches pour mieux comprendre les rôles complexes du TNF-α dans la SLA et affiner les stratégies thérapeutiques (Guidotti, 2021). Bien que l'inhibition du TNF-α reste une piste potentielle de traitement, des études supplémentaires sont essentielles pour déterminer les méthodes les plus efficaces pour cibler cette voie inflammatoire dans la SLA.
En conclusion, le TNF-α joue un rôle central dans la neuroinflammation et la neurodégénérescence dans des maladies telles que la MA, la MP et la SLA. Malgré les différences dans leurs mécanismes sous-jacents, les processus inflammatoires induits par le TNF-α semblent être un facteur commun dans la progression de la maladie. Alors que les thérapies ciblant le TNF-α sont prometteuses dans les modèles précliniques, les résultats cliniques ont été incohérents, soulignant la nécessité d'une meilleure compréhension du rôle du TNF-α. Les recherches futures devraient se concentrer sur le perfectionnement des traitements ciblés qui modulent la signalisation du TNF-α, offrant potentiellement des thérapies pour ralentir ou arrêter la progression de ces maladies neurodégénératives.
Quelles preuves suggèrent que la modulation du TNF-α peut ralentir ou prévenir les maladies neurodégénératives ?
Étant donné son rôle central dans la neuroinflammation, le TNF-α est essentiel à la progression de nombreuses maladies neurodégénératives, notamment la MA, la MP et la SLA. L'élévation chronique du TNF-α dans le SNC entraîne des lésions neuronales et une accumulation de protéines toxiques, qui contribuent ensemble à la progression de la maladie. À ce titre, la modulation du TNF-α est apparue comme une stratégie prometteuse pour ralentir, voire stopper la progression de ces maladies.
Les traitements anti-TNF-α, efficaces dans le traitement de diverses maladies auto-immunes et inflammatoires, ont attiré l'attention en raison de leur potentiel dans les maladies neurodégénératives (Gonzalez Caldito, 2023). Des médicaments tels que l'étanercept (Enbrel®) et l'infiximab (Remicade®), largement utilisés dans le traitement de maladies telles que la polyarthrite rhumatoïde et d'autres maladies inflammatoires périphériques, ont une efficacité limitée dans les troubles du SNC en raison d'une mauvaise pénétration de la BHE (Decourt, 2017). D'autres inhibiteurs du TNF-α, dont l'adalimumab (Humira®), le golimumab (Simponi®) et le certolizumab pegol (Cimzia®), sont confrontés à des difficultés similaires pour accéder au SNC.
Ce défi de l'inhibition du TNF-α dans les maladies du SNC est aggravé par le risque d'effets secondaires graves, tels qu'une susceptibilité accrue aux infections et aux maladies démyélinisantes (Kemanetzoglou, 2017). Par exemple, dans la sclérose en plaques, des traitements comme le lenercept ont aggravé l'évolution de la maladie (Maguire, 2021). Les doubles rôles du TNF-α dans la neuroprotection et l'inflammation soulignent la nécessité d'un ciblage plus sélectif. Les recherches sur le TNFR1 et le TNFR2 ont mis en évidence que le ciblage sélectif du TNFR1, tout en épargnant le TNFR2, pourrait offrir une approche plus sûre et plus efficace pour traiter la sclérose en plaques (Fresegna, 2020).
En réponse à ces limites, le développement d'inhibiteurs sélectifs du TNF-α qui ciblent spécifiquement le TNFR1 pour une utilisation dans le SNC, tels que l'atrosab et l'atrosimab, a donné des résultats prometteurs dans des modèles précliniques de neuroinflammation, tels que l'encéphalomyélite auto-immune expérimentale (EAE) (Richter, 2021). De même, un essai de phase 1b pour le XPro1595, un inhibiteur du TNF-α sélectif pour le TNF-α soluble, a montré des résultats prometteurs dans la MA (NCT03943264). Ces thérapies visent à offrir une approche plus ciblée, améliorant la sécurité et l'efficacité pour les maladies neurodégénératives.
Bien que les résultats précliniques soient prometteurs, des essais cliniques supplémentaires sont nécessaires pour établir l'innocuité et l'efficacité de ces inhibiteurs sélectifs du TNF-α chez l'homme. Ces études seront cruciales pour déterminer si ces thérapies peuvent apporter des bénéfices aux patients atteints de maladies neurodégénératives. À mesure que la recherche se poursuit, le développement de traitements ciblant le TNF-α dans le SNC pourrait offrir une approche importante pour ralentir ou prévenir la neurodégénérescence, améliorer les résultats pour les patients et faire progresser le domaine du traitement de la neuroinflammation (Decourt, 2017; Hampel, 2020; Zahedipour, 2022).
Notre équipe se fera un plaisir de répondre à toutes vos questions sur la fonction du TNF-α dans la microglie et sa contribution aux maladies neurodégénératives ou de vous fournir des informations spécifiques sur les modèles de la MA, de la SLA et de la MP que nous utilisons pour les études d'efficacité thérapeutique.
En savoir plus sur nos modèles de maladies neurodégénératives
Contenu connexe
Informations actualisées sur le TNF-α et la microglie dans les maladies neurodégénératives et les meilleures pratiques liées à l'évaluation des agents thérapeutiques dans les modèles animaux de maladies neurodégénératives.
Le TNF-α et les astrocytes dans les maladies neurodégénératives
Présentation générale de la signalisation du TNF-α dans les astrocytes, de son rôle dans la neurodégénérescence et des stratégies thérapeutiques ciblant cette voie.
Microglie, astrocytes et protéine tau dans les maladies neurodégénératives
Comment la neuroinflammation induite par les cellules gliales favorise l'agrégation et la propagation de la protéine tau ainsi que la perte neuronale dans la maladie d'Alzheimer et d'autres tauopathies.
Microglie, astrocytes et α-synucléine dans la maladie de Parkinson
Comment l'α-synucléine influence les microglies et les astrocytes dans la maladie de Parkinson et d'autres synucléinopathies.
Dysfonctionnement mitochondrial dans les microglies et les astrocytes
Le rôle du dysfonctionnement mitochondrial dans les microglies et les astrocytes dans les maladies neurodégénératives, notamment la maladie d'Alzheimer, la maladie de Parkinson et la SLA.
Sénescence microgliale et maladies neurodégénératives
Cette ressource fournit un aperçu de la sénescence microgliale et de son rôle dans les maladies neurodégénératives, notamment la maladie d'Alzheimer (MA) et la maladie de Parkinson (MP).
Morphologie de la microglie dans la SLA, la maladie d'Alzheimer et la maladie de Parkinson
Une vue d'ensemble de l'analyse morphologique des microglies et des applications à la recherche sur les maladies neurodégénératives et à la découverte et au développement de médicaments.
Amyloïde-β et microenvironnement inflammatoire chez la souris Alzheimer
Nous avons analysé les relations spatiales complexes entre les plaques β-amyloïdes, la microglie activée et au repos, et les astrocytes dans un modèle transgénique APP/PS1.