什么是TDP-43?
转录反应DNA结合蛋白(TDP-43)是一种高度保守的蛋白质,能够结合RNA和DNA。该蛋白由TARDBP基因编码,属于细胞核内异质核核糖核蛋白(hnRNP)家族(Cohen, 2011;de Boer, 2020;Jo, 2020;Corbet, 2021;Tziortzouda, 2021)。
在正常生理条件下,TDP-43主要存在于细胞核内,但也会在核与细胞质之间进行活跃转运。当TDP-43异常定位至细胞质时,可能引发肌萎缩侧索硬化症(ALS)和额颞叶痴呆(FTD)等蛋白质病。在这些病理状态下,它会形成有害的聚集物,从而促进神经退行性变(de Boer, 2020;Jo, 2020;Chhangani, 2021;Scialo, 2025)。
TDP-43的功能
TDP-43在调控细胞核内的基因表达和RNA加工过程中发挥关键作用。
TDP-43的核内功能包括(de Boer, 2020;Corbet, 2021;Dykstra, 2025):
- 通过抑制剪接过程中隐性外显子的包含来调控RNA转录
- mRNA的维持、代谢与转运
- 微RNA(miRNA)的成熟
TDP-43可迁移至细胞质执行不同功能,其活动受负反馈机制调控。
TDP-43的胞质功能包括(Cohen, 2011;Jo, 2020;Corbet, 2021;Tziortzouda, 2021):
- mRNA的稳定性与翻译
- 两种结构的形成:
- 应激颗粒——对神经元抵御氧化应激至关重要
- 核糖核蛋白颗粒——对mRNA转运和miRNA生物合成至关重要
此外,TDP-43与线粒体基因组结合,参与呼吸链相关通路。该蛋白对胚胎发育早期中枢神经细胞的正常发育至关重要。事实上,在小鼠模型中,TDP-43功能缺失已被证实会导致胚胎致死(Cohen, 2011;de Boer, 2020)。
TDP-43的多样化功能源于其不同结构域(Cohen, 2011;de Boer, 2020;Jo, 2020;Tziortzouda, 2021;Corbet, 2021):
- N端结构域(NTD)促进TDP-43的自寡聚化。该结构域还包含核定位信号(NLS),对蛋白质转运至细胞核至关重要。
- RNA识别基序(RRM1和RRM2)负责结合核酸,包括RNA和DNA。
- 核输出信号(NES)促进TDP-43的转运。
- C端结构域(CTD)调节蛋白质溶解度,介导病理性聚集,并参与将TDP-43招募至应激颗粒。
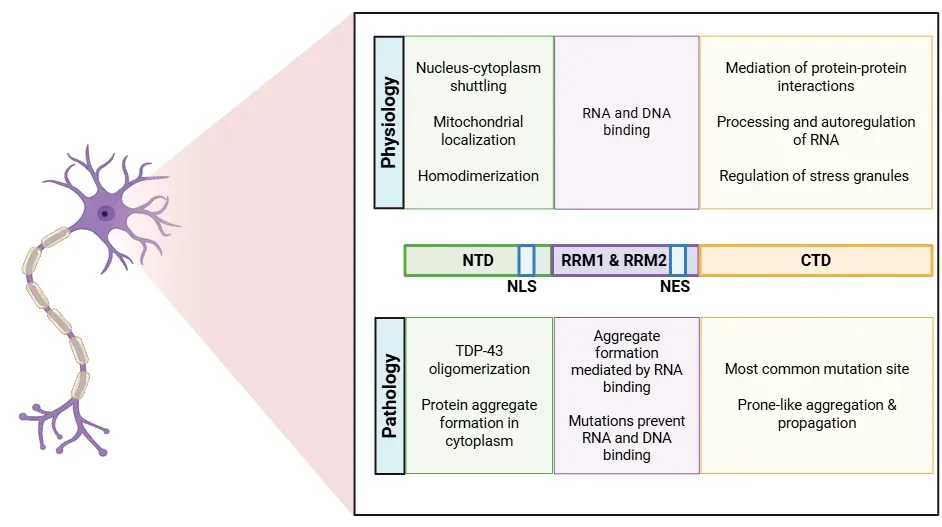
TDP-43蛋白的结构,详细阐述每个区域的生理和病理作用。
TDP-43病理在肌萎缩侧索硬化症(ALS)和额颞叶痴呆(FTD)中的意义是什么?
TDP-43蛋白病变是包括ALS和FTD在内的多种神经退行性疾病的标志性病理特征。TDP-43蛋白的聚集与疾病进展及严重程度密切相关。在20-50%的阿尔茨海默病(AD)患者中也可发现TDP-43聚集物,这些聚集物与记忆功能下降及脑萎缩加剧相关(Prasad, 2019)。
TDP-43蛋白病相关的致病过程包含毒性获得功能和丧失功能两种机制。TDP-43的裂解、过度磷酸化及泛素化修饰会导致其在细胞核和细胞质中形成毒性聚集物(Mackenzie, 2008;Xu, 2014;Yang, 2014;Scotter, 2015;Meneses, 2021)。
TDP-43病理学中已发现多种基因突变,其中多数定位于CTD区域。例如,基因剪接异常导致隐性外显子被纳入,进而引发核内TDP-43缺失,从而诱发TDP-43病理。受此类突变影响的两个相关基因包括:
- UNC13A:功能性TDP-43缺失导致UNC13A mRNA中隐性外显子被纳入,进而降低对突触传递至关重要的UNC13A蛋白表达(Garcia-Montojo, 2024)。
- Stathmin2 (STMN2):该基因对运动神经元生长与修复至关重要。TDP-43功能缺失会导致隐性外显子纳入及多聚腺苷酸化,进而引发STMN2基因剪接异常与表达耗竭(Suk, 2020;Ma, 2022)。
其他与TDP-43病理相关的显著突变涉及C9orf72基因(关联经典ALS和FTD)、TARDBP及ALS2基因(de Boer, 2020;Jo, 2020)。
在病理状态下,TDP-43通过多种机制持续从细胞核向细胞质转运(Yang, 2014;Prasad, 2019;Suk, 2020;Garcia-Montojo, 2024):
- NTD内核定位信号(NLS)突变。
- 应激颗粒调控失常:氧化应激或热休克后,TDP-43、RNA及其他蛋白质在胞质中异常堆积。生理状态下,细胞应激消退后TDP-43通常会重新转运至细胞核。
- 核孔复合体功能障碍:(G4C2)30RNA水平降低会减少Ran蛋白表达,而Ran蛋白调控着TDP-43的核定位。其他蛋白如Nup62和Kpnb1也参与该转运过程。
- 胞质聚集物:TDP-43在胞质中形成聚集物会引发恶性循环,进一步破坏核质运输和核孔复合体功能,导致更多TDP-43蛋白从细胞核中逸出。
这些病理变化导致:
- 功能丧失(Yang, 2014;de Boer, 2020;Suk, 2020;Garcia-Montojo, 2024):
- 毒性功能获得(Xu, 2014;Prasad, 2019;Suk, 2020;Meneses, 2021):
- 病理性聚合体的形成导致蛋白质聚集增加和神经毒性
- 通过外泌体或突触传递的朊病毒样传播。小胶质细胞、星形胶质细胞和少突胶质细胞可吞噬TDP-43并将其传递至神经元
- 最终,TDP-43聚集体的存在导致神经元死亡
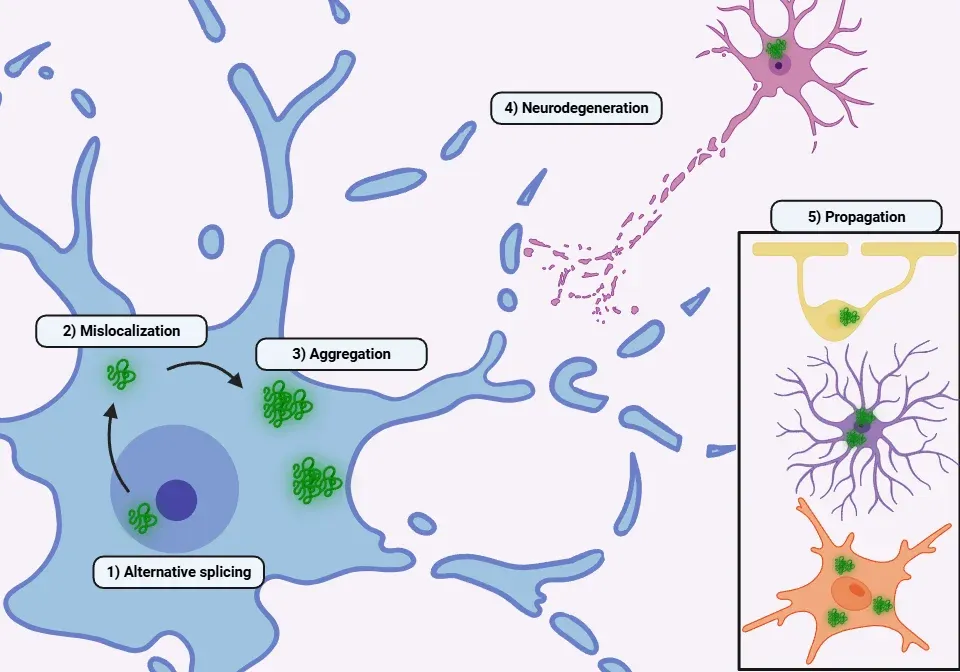
TDP-43蛋白病理的病理生理机制综述
肌萎缩侧索硬化症(ALS)
TDP-43的胞质聚集被认为是ALS的主要病理标志(Xu, 2014;Scotter, 2015;Prasad, 2019;de Boer, 2020;Suk, 2020;Hu, 2024):
- 超过90%的散发性及家族性ALS患者存在TDP-43聚集物。
- 由SOD1或 FUS基因突变引发的ALS通常不表现TDP-43病理特征,而TARDBP基因突变则会导致TDP-43病理改变,并直接引发家族性ALS。
- 然而,TARDBP突变仅存在于约4%的家族性ALS病例中,而家族性ALS病例占所有ALS病例的比例不足10%。
- 其他与ALS中TDP-43聚集相关的基因突变包括A90V(位于核定位信号区域)、G294V和A315T。
- 根据TDP-43病理的具体位置,可能出现执行功能障碍、语言困难及行为异常等症状。
目前已确认ALS中存在三种TDP-43病理类型(de Boer, 2020):
- 混合型神经元-胶质细胞病理
- 胶质病理(尤其星形胶质细胞)
- 神经元病理
TDP-43病理在皮层第五层神经元和脊髓运动神经元中最为显著。ALS患者组织显示TDP-43沉积存在区域性差异(Scotter, 2015;Wu, 2024):
- C端截短片段在脑组织中富集
- 脊髓包涵体中以全长TDP-43为主
小鼠模型中TDP-43功能的部分丧失足以引发神经退行性变、进行性运动功能障碍、瘫痪直至死亡(Xu, 2014;Yang, 2014)。
FTD
FTD(病理学上称为额颞叶变性症FTLD)的特征在于神经元和胶质细胞中存在TDP-43、tau或FUS蛋白聚集物(Prasad, 2019;Ho, 2024;Hu, 2024)。
- TDP-43突变仅与少数额颞叶痴呆病例相关。
- 例如,C9orf72突变约占 FTD 病例的 13%。
- 然而,高达 50% 的 FTD 患者体内存在 TDP-43 聚集体。
- 另一个重要的突变是GRN基因(前蛋白),它会导致功能丧失,并以常染色体显性方式遗传。
与TDP-43相关的FTD症状包括人格、行为及语言能力的改变,例如丧失同理心、不当社交行为及沟通困难(Prasad, 2019;de Boer, 2020;Jo, 2020)。
参见资源:额颞叶痴呆的神经影像学与临床试验
FTD患者的血清TDP-43总水平通常显著低于健康对照组,尤其在C9orf72重复序列扩张携带者或伴有运动神经元疾病表型的FTD患者中更为明显。这种水平下降被推测源于该蛋白被隔离至不溶性聚集物中(Katisko, 2022)。
FTLD病理学分为三类:FTLD-tau(35-50%)、FTLD-FUS(10%)及FTLD-TDP(约50%)。其中FTLD-TDP进一步细分为以下亚型(Prasad, 2019;de Boer, 2020;Meneses, 2021):
- A型:紧凑或新月形神经元胞质内包涵体(NCI),通常见于新皮层。
- B型:弥漫性或颗粒状NCI,常见于皮质及皮质下少突胶质细胞,并伴随运动神经元疾病。
- C型:大量营养不良性神经突,主要见于新皮质。
- D型:大量神经元胞核内包涵体。
参见我们的创新成果:扩散MRI与额颞叶痴呆(FTD) 及MRI脑萎缩
TDP-43是否是潜在的治疗靶点?
当前治疗策略主要侧重三种方法:清除毒性蛋白质、恢复其功能,以及应对其耗竭引发的下游效应。
载体化抗体
- ACI-5891是一种通过AAV9递送的载体化单克隆抗体,专门针对病理性TDP-43。在ALS和FTD小鼠模型中,单次脑室内注射成功将病理性磷酸化TDP-43信号降低高达68%(Val, 2025)。
- 抗体3B12A可检测错误折叠TDP-43聚集物中E246和D247残基的异常暴露,经工程改造后能促进TDP-43经蛋白酶体降解(Francois-Moutal, 2021)。
- VH7Vk9通过靶向RRM1结构域,促进蛋白酶体或自噬介导的降解(Francois-Moutal, 2021)。
小分子
- 黄芩苷作为结构矫正剂,能将现有错误折叠蛋白转化为功能性形式(Chang, 2024)。
- M102是核因子红细胞2相关因子2(NRF2)和热休克因子1(HSF-1)通路的激活剂,在ALS动物模型中通过减轻氧化应激和TDP-43聚集效应展现出潜力(San Gil, 2025)。
- 其他分子已在果蝇模型中得到探索(Francois-Moutal, 2021):
- rTRD0,靶向RRM1核酸结合界面
- nTRD22,靶向N端结构域以促进TDP-43清除
反义寡核苷酸(ASO)
- QRL-201靶向STMN2(TDP-43病理中发生剪接异常的蛋白质),有助于改善运动神经元功能。该疗法目前正针对散发性及C9orf72型ALS开展I期临床试验(Liu, 2024)。
- Qalsody获FDA批准用于治疗携带SOD1突变的ALS患者,但不针对TDP-43病理(San Gil, 2025)。
基因疗法
- 针对UNC13a等受累基因的异常剪接,相关预临床开发项目正在推进(San Gil, 2025)。
分子伴侣靶向疗法
- 在细胞模型中过度表达J域蛋白(DNAJB1、DNAJB2a、DNAJB4、DNAJB5)可显著降低不溶性TDP-43水平(San Gil, 2025)。
14-3-3θ
- 在ALS小鼠模型中靶向错误折叠的TDP-43,改善行为缺陷(San Gil, 2025)。
自噬诱导
- 可增强蛋白质聚集物的清除效率(Hayes, 2022):
- 雷帕霉素等mTOR抑制剂
- TFEB诱导剂(如海藻糖和依布地拉斯特)
- 参见资源:神经退行性疾病中微胶质细胞自噬缺陷&自噬与转录因子EB(TFEB)
C端甘氨酸富集区与RRM2结构域对驱动相分离和聚集至关重要,使其成为特异性抗体的理想靶点(Riemenschneider, 2023)。正如作者所述,靶向关键结构域需谨慎操作——部分N端肽段的活性免疫接种导致小鼠死亡,这可能源于TDP-43在细胞核内的生理功能(Riemenschneider, 2023)。
多个研究团队已在TDP-43 δNLS ALS小鼠模型中通过治疗干预实现疾病修饰,重要发现包括:
- AIT-101:一种抑制PIKfyve的小分子药物。该治疗可减轻体重下降、改善运动功能缺陷、降低血浆及脑脊液中神经丝轻链(NfL)水平,并减少TDP-43聚集物(Young, 2023)。
- VRG50304:该化合物可降低血浆及脑脊液中NfL水平,同时减少致病皮层的酸性鞘磷脂酶活性(Stomakhina, 2021)。
- AAV9/NF242:脑室内注射Rho鸟苷酸交换因子N端片段可同时改善小鼠寿命和运动功能(Droppelmann, 2024)。
详见资源库:TDP-43 ΔNLS (rNLS8) 小鼠在ALS药物研发中的应用
尽管多种靶向TDP-43的疗法在ALS临床试验中失败(如arimoclomol、苯丁酸钠、熊去氧胆酸牛磺酸、秋水仙碱及BIIB105,San Gil, 2025),TDP-43仍持续作为重要靶点推进研究。尽管目前尚未有基于TDP-43的疗法在人体试验中成功延缓疾病进展,但众多疗法仍处于临床前研发阶段,为未来进展奠定了坚实基础。要实现有效治疗,需采取创新策略同时解决TDP-43聚集与功能丧失问题,从而为ALS治疗的潜在突破铺平道路(San Gil, 2025)。
我们的团队很乐意解答关于TDP-43的任何疑问,或提供有关我们用于治疗有效性研究的模型的具体信息。
相关内容
关于多发性硬化症的最新信息,以及在MS动物模型中评估治疗药物的最佳实践。
小胶质细胞与神经元相互作用与神经退行性疾病
对小胶质细胞与神经元之间直接相互作用的简要综述,以及这些细胞间相互作用在神经退行性疾病中可能受到的影响。
ALS小鼠模型用于药物研发
指导如何最有效地使用肌萎缩侧索硬化症(ALS)的实验动物模型(小鼠和大鼠模型)进行临床前治疗测试。
TDP-43 ΔNLS (rNLS8) 小鼠用于ALS药物研发
该资源提供了有关使用ΔNLS(deltaNLS、hTDP-43ΔNLS、hTDP-43DeltaNLS、dNLS、TDP43 NLS、rNLS8)TDP-43 ALS转基因小鼠模型进行临床前治疗研究的信息。
额颞叶痴呆症和临床试验中的神经影像学
核磁共振成像和正电子发射断层扫描成像生物标记物在了解额颞叶痴呆(FTD)变体方面的效用,以及它们在FTD临床试验中的终点用途。