TDP-43 ― 筋萎縮性側索硬化症(ALS)および前頭側頭型認知症(FTD)におけるその役割
TDP-43の概要、その生理的役割、ALSおよびFTD病理における意義、ならびにTDP-43を標的とした治療戦略について。
この資料では以下について説明しております:
TDP-43とは何でしょうか?
転写調節DNA結合タンパク質(TDP-43)は、RNAおよびDNAに結合する高度に保存されたタンパク質です。TARDBP遺伝子によってコードされ、核内における異種核リボ核タンパク質(hnRNP)ファミリーに属します(Cohen, 2011;de Boer, 2020;Jo, 2020;Corbet, 2021;Tziortzouda, 2021)。
正常な生理状態では、TDP-43は主に核内に存在しますが、核と細胞質の間を活発に行き来しています。細胞質へ異常局在化したTDP-43は、筋萎縮性側索硬化症(ALS)や前頭側頭型認知症(FTD)などのタンパク質病を引き起こす可能性があります。これらの病態において、TDP-43は有害な凝集体を形成し、神経変性に寄与します(de Boer, 2020;Jo, 2020;Chhangani, 2021;Scialo, 2025)。
TDP-43 の機能
TDP-43 は、核内における遺伝子発現および RNA 処理の調節に重要な役割を果たしています。
TDP-43の核内機能には以下が含まれます(de Boer, 2020;Corbet, 2021;Dykstra, 2025):
- スプライシング過程における隠れたエクソンの包含を抑制することによるRNA転写調節
- mRNAの維持、代謝、輸送
- マイクロRNA(miRNA)の成熟
TDP-43は細胞質に移動して異なる機能を果たし、負のフィードバック機構によって調節されています。
TDP-43の細胞質における機能には以下のものが含まれます(Cohen, 2011;Jo, 2020;Corbet, 2021;Tziortzouda, 2021):
- mRNAの安定性と翻訳
- 二つの構造体の形成:
- 酸化ストレスから神経細胞を保護するために不可欠なストレス顆粒
- リボ核タンパク質顆粒(mRNA輸送およびmiRNA生合成に重要)
さらに、TDP-43はミトコンドリアゲノムと結合し、呼吸鎖に関連する経路に関与しています。このタンパク質は、胚発生の初期段階における中枢神経細胞の正常な発達に極めて重要です。実際、マウスモデルでは、TDP-43機能の喪失が胚致死性であることが示されています(Cohen, 2011;de Boer, 2020)。
TDP-43の多様な機能は、その様々な領域に由来しています(Cohen, 2011;de Boer, 2020;Jo, 2020;Tziortzouda, 2021;Corbet, 2021):
- N末端ドメイン(NTD)はTDP-43の自己オリゴマー化を促進します。このドメインには核局在シグナル(NLS)も含まれており、タンパク質を核へ輸送する上で重要です。
- RNA認識モチーフ(RRM1およびRRM2)は、RNAとDNAの両方を含む核酸への結合を担っています。
- 核輸出シグナル(NES)は、TDP-43の輸送を促進します。
- C末端ドメイン(CTD)は、タンパク質の可溶性を調節し、病理学的凝集を媒介するほか、ストレス顆粒へのTDP-43の動員にも寄与します。
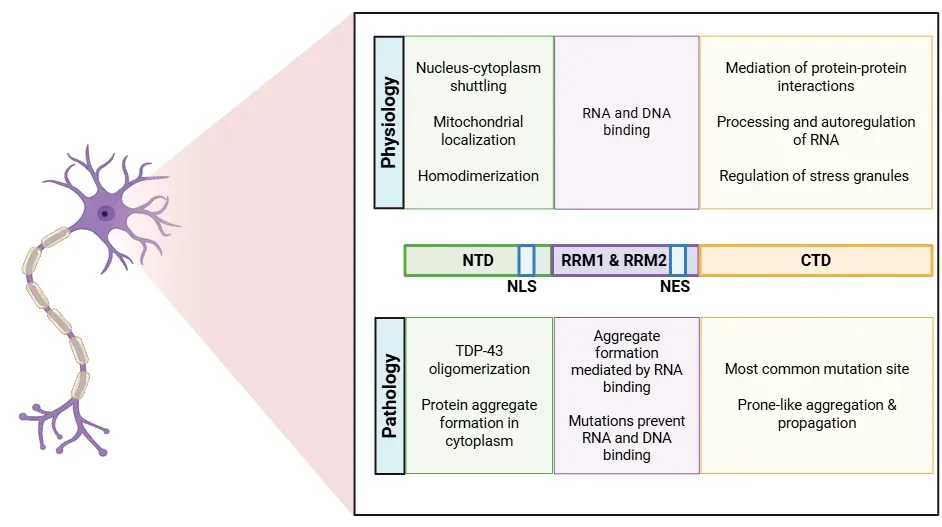
TDP-43タンパク質の構造、および各部位の生理的・病理的役割の詳細について。
ALSおよびFTDにおけるTDP-43病理の意義とは何でしょうか?
TDP-43タンパク質病変は、ALSやFTDを含む複数の神経変性疾患の特徴的な病理所見です。TDP-43タンパク質の凝集は、疾患の進行や重症度と密接に関連しています。また、TDP-43凝集体はアルツハイマー病(AD)患者の20~50%にも認められ、記憶機能の低下や脳萎縮の進行と相関関係が報告されています(Prasad, 2019)。
TDP-43タンパク質病変に関連する病原性プロセスには、毒性のある機能獲得メカニズムと機能喪失メカニズムの両方が含まれます。TDP-43の切断、過剰リン酸化、ユビキチン化は、核内および細胞質の両方で毒性凝集体の形成を引き起こします(Mackenzie, 2008;Xu, 2014;Yang, 2014;Scotter, 2015;Meneses, 2021)。
TDP-43病理においては様々な遺伝子変異が報告されており、その多くはCTD領域に局在しています。例えば、遺伝子誤スプライシングによる核内TDP-43の喪失が、隠れたエクソンの包含を引き起こし、TDP-43病理が生じる可能性があります。これらの変異の影響を受ける関連遺伝子として、以下の2つが挙げられます:
- UNC13A:機能的なTDP-43の喪失は、UNC13A mRNAにおける隠れたエクソンの包含を引き起こし、シナプス伝達に重要なUNC13Aタンパク質の発現低下につながります(Garcia-Montojo, 2024)。
- Stathmin2 (STMN2): STMN2は運動ニューロンの成長と修復に重要です。TDP-43機能の喪失は、隠れたエクソンの包含とポリアデニル化により、STMN2の誤ったスプライシングと減少を引き起こします(Suk, 2020;Ma, 2022)。
TDP-43病理に関連するその他の重要な変異には、C9orf72遺伝子(古典的ALSおよびFTDと関連)、TARDBP遺伝子、ALS2遺伝子などが挙げられます(de Boer, 2020;Jo, 2020)。
病理学的状況下では、TDP-43はいくつかのメカニズムを通じて核から細胞質へ増加して輸送されます(Yang, 2014;Prasad, 2019;Suk, 2020;Garcia-Montojo, 2024)。
- NTD内のNLSにおける変異。
- ストレス顆粒の調節機能の障害:酸化ストレスや熱ショック後、TDP-43、RNA、その他のタンパク質が細胞質に蓄積します。生理的条件下では、細胞ストレスが解消されると、TDP-43は通常、核へ再移行します。
- 核膜孔複合体の機能障害:(G4C2)30RNAレベルの減少により、TDP-43の核局在を調節するRanの発現が低下します。Nup62やKpnb1などの他のタンパク質も、この輸送過程に関与しています。
- 細胞質凝集体:TDP-43の細胞質凝集体形成は悪循環を引き起こし、核細胞質輸送と核孔複合体をさらに破壊します。これにより、より多くのTDP-43が核外へ流出します。
これらの病理学的変化は以下をもたらします:
- 機能喪失(Yang, 2014;de Boer, 2020;Suk, 2020;Garcia-Montojo, 2024):
- RNA代謝の調節異常
- 核内TDP-43の減少
- オートファジーの障害
- ミトコンドリア機能障害
- 毒性獲得機能(Xu, 2014;Prasad, 2019;Suk, 2020;Meneses, 2021):
- 病理学的ポリマーの形成によるタンパク質凝集の増加と神経毒性の増大
- エクソソームまたはシナプス伝達を介したプリオン様伝播。ミクログリア、アストロサイト、オリゴデンドロサイトはTDP-43を貪食し、神経細胞へ伝達することが可能です
- 関連リソース:ミクログリア-ニューロン相互作用と神経変性疾患
- 最終的に、TDP-43凝集体の存在が神経細胞死に寄与します
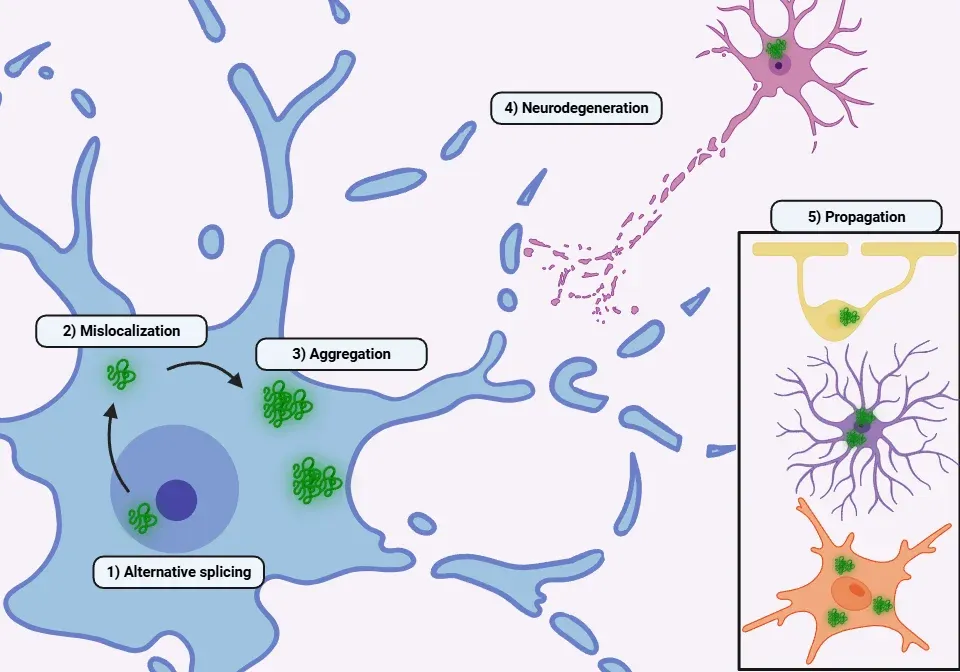
TDP-43タンパク質病の病態生理学的メカニズムの概要
筋萎縮性側索硬化症(ALS)
TDP-43の細胞質凝集は、ALSの主要な病理学的特徴と考えられています(Xu, 2014;Scotter, 2015;Prasad, 2019;de Boer, 2020;Suk, 2020;Hu, 2024):
- 散発性および家族性ALS患者の90%以上がTDP-43凝集体を示します。
- 一方、SOD1またはFUSの変異によるALSでは通常TDP-43病理は認められませんが、TARDBPの変異はTDP-43病理を引き起こし、家族性ALSの直接的な原因となります。
- しかしながら、TARDBP変異は家族性ALS症例の約4%にしか認められず、これは全ALS症例の10%未満に相当します。
- ALSにおけるTDP-43凝集に関連するその他の遺伝子変異には、A90V(NLS領域に位置)、G294V、A315Tなどが含まれます。
- TDP-43病変の発生部位によって、実行機能障害、言語障害、行動異常などの症状が現れることがあります。
ALSでは3種類のTDP-43病理が同定されています(de Boer, 2020):
- 混合型(神経細胞とグリア細胞の病変)
- グリア病変(特にアストロサイトにおけるもの)
- ニューロン病変
TDP-43病変は、皮質第V層ニューロンおよび脊髄運動ニューロンにおいて最も顕著です。ALS患者の組織では、沈着するTDP-43の種に部位ごとの差異が認められます(Scotter, 2015;Wu, 2024):
- C末端切断断片は脳内で豊富に認められます
- 脊髄内包体では全長TDP-43が優勢です
マウスモデルにおいてTDP-43機能の部分的喪失は、神経変性、進行性運動機能障害、麻痺、そして最終的には死を引き起こすのに十分です(Xu, 2014;Yang, 2014)。
参照:筋萎縮性側索硬化症(ALS)のTDP-43マウスモデル
FTD(
)FTD(前頭側頭型認知症)は、病理学的観点からは前頭側頭葉変性症(FTLD)とも呼ばれ、ニューロンおよびグリア細胞内にTDP-43、タウ、またはFUSタンパク質の凝集体が存在することが特徴です(Prasad, 2019;Ho, 2024;Hu, 2024)。
- TDP-43変異は、FTD症例のわずか一部に関連しています。
- 例えば、C9orf72変異はFTD症例の約13%を占めています。
- しかし、TDP-43凝集体は、FTD患者の最大50%に見られます。
- もう一つの重要な変異はGRN遺伝子(プログラニュリン)であり、これは機能喪失を引き起こし、常染色体優性遺伝します。
TDP-43に関連するFTDの症状には、共感性の喪失、不適切な社会的行動、コミュニケーション困難など、人格、行動、言語能力の変化が含まれます(Prasad, 2019;de Boer, 2020;Jo, 2020)。
FTD患者では、特にC9orf72反復拡張保因者や運動ニューロン疾患を併発するFTD患者において、血清中TDP-43総量が健常者と比べて著しく低い傾向があります。この減少は、タンパク質が不溶性凝集体に封じ込められることに起因すると考えられています(Katisko, 2022)。
FTLDでは3種類の病理が報告されています:FTLD-tau(35-50%)、FTLD-FUS(10%)、FTLD-TDP(約50%)。FTLD-TDPはさらに以下のサブタイプに分類されます(Prasad, 2019;de Boer, 2020;Meneses, 2021):
- タイプA:通常新皮質に認められる、緻密または三日月状の神経細胞質封入体(NCI)。
- タイプB:拡散性または顆粒状のNCI。皮質および皮質下オリゴデンドロサイトに多く認められ、運動ニューロン疾患と関連します。
- タイプC:主に新皮質に認められる多数のジストロフィックニューリト。
- タイプD:多数の神経細胞核内封入体。
当社のイノベーションをご覧ください:拡散MRIと前頭側頭型認知症(FTD)および前頭側頭型認知症(FTD)およびMRI脳萎縮
TDP-43は治療標的となり得るのでしょうか?
現在の治療戦略では、主に三つのアプローチが重視されています:毒性タンパク質の除去、その機能の回復、そしてその減少による下流への影響への対処です。
ベクター化抗体
- ACI-5891は、AAV9を用いて送達されるベクター化モノクローナル抗体であり、病理学的TDP-43を特異的に標的とします。ALSおよびFTDのマウスモデルにおいて、単回脳室内投与により病理学的リン酸化TDP-43シグナルを最大68%まで減少させることに成功しました(Val, 2025)。
- 抗体 3B12A は、誤って折りたたまれた TDP-43 凝集体における E246 および D247 残基の異常な露出を検出し、TDP-43 のプロテアソームによる分解を促進するように設計されています(Francois-Moutal、2021)。
- VH7Vk9 は、RRM1 ドメインに焦点を当てることによって、プロテアソームまたはオートファジーによる分解を促進します(Francois-Moutal、2021)。
低分子
- バイカレインは、構造修正剤として作用し、既存の誤って折りたたまれたタンパク質を機能的な形態に変換するのに役立ちます(Chang、2024)。
- M102 は、核因子エリスロイド 2 関連因子 2 (NRF2) および熱ショック因子 1 (HSF-1) 経路の活性化因子であり、酸化ストレスと TDP-43 凝集の影響を軽減することで、ALS の動物モデルにおいて有望な結果を示しています(San Gil、2025)。
- ハエのモデルでは、さらに他の分子も研究されています(Francois-Moutal、2021年)。
- RRM1 核酸結合インターフェースを標的とする rTRD0
- nTRD22:N 末端ドメインを標的とし、TDP-43 のクリアランスを促進
アンチセンスオリゴヌクレオチド(ASO)
- QRL-201 は、TDP-43 病態で誤ってスプライシングされるタンパク質である STMN2 を標的とし、運動ニューロンの機能改善に役立ちます。この治療法は現在、散発性および C9orf72-ALS に対する第 1 相試験が行われています(Liu、2024)。
- QalsodyはSOD1変異を伴うALS患者に対してFDA承認を受けておりますが、TDP-43病理には対応しておりません(San Gil, 2025)。
遺伝子治療
- 前臨床開発段階では、UNC13aなどの他の影響を受ける遺伝子の異常スプライシングを修正する治療法が検討されています(San Gil, 2025)。
シャペロン標的療法
- Jドメインタンパク質(DNAJB1、DNAJB2a、DNAJB4、DNAJB5)を過剰発現させることで、細胞モデルにおいて不溶性TDP-43を大幅に減少させることが確認されています(San Gil, 2025)。
14-3-3θ
- ALSマウスモデルにおいて、誤って折りたたまれたTDP-43を標的とし、行動障害の改善につながります(San Gil, 2025)。
オートファジー誘導
- タンパク質凝集体の除去を促進します(Hayes, 2022):
- ラパマイシンなどのmTOR阻害剤
- トレハロースやイブディラストなどのTFEB誘導剤
- 関連リソースをご覧ください:神経変性疾患におけるミクログリアのオートファジー障害&オートファジーと転写因子EB(TFEB)
C末端グリシン豊富な領域およびRRM2ドメインは、相分離と凝集を促進する上で極めて重要であり、特異的抗体の標的として有望です(Riemenschneider, 2023)。著者らが述べているように、必須ドメインを標的とする際には注意が必要です。一部のN末端ペプチドを用いた能動免疫は、おそらく核内におけるTDP-43の生理的役割に起因して、マウスに致死性を引き起こしました(Riemenschneider, 2023)。
複数の研究グループが、TDP-43 δNLS ALSマウスモデルにおける治療的介入による疾患修飾効果を実証しています。主な知見は以下の通りです:
- AIT-101:PIKfyveを阻害する低分子化合物です。本治療により、体重減少の抑制、運動機能障害の改善、血漿および脳脊髄液(CSF)中の神経フィラメント軽鎖(NfL)レベルの低下、ならびにTDP-43凝集体の減少が確認されています(Young, 2023)。
- VRG50304:本化合物は血漿および髄液中のNfLレベルを低下させるとともに、皮質における酸性スフィンゴミエリン分解酵素活性を減少させることが確認されています(Stomakhina, 2021)。
- AAV9/NF242:ローグアノシンヌクレオチド交換因子のN末端断片を脳室内投与した結果、マウスの寿命と運動能力の両方が改善されました(Droppelmann, 2024)。
詳細については、当社のリソース「ALS治療薬開発のためのTDP-43 ΔNLS (rNLS8) マウス」をご参照ください。
アリモクロモール、フェニル酪酸ナトリウム、ウルソドキソコリチン、コルヒチン、BIIB105など、TDP-43を標的とした複数の治療法がALS臨床試験で失敗に終わりましたが(San Gil, 2025)、TDP-43は現在進行中の研究において重要な標的であり続けています。現時点では、ヒトにおいて疾患進行を遅らせる能力を実証したTDP-43ベースの治療法はまだ存在しませんが、多くの治療法が前臨床開発段階にあり、将来の進展に向けた強固な基盤を提供しています。効果的な治療法への進展には、TDP-43の凝集とその機能喪失の両方に対処する革新的な戦略が必要であり、それがALS治療における潜在的なブレークスルーへの道を開くことでしょう(San Gil, 2025)。
当チームは、TDP-43に関するご質問や、治療効果研究に使用するモデルに関する具体的な情報について、喜んでお答えいたします。
関連コンテンツ
多発性硬化症に関する最新情報ならびに、MS動物モデルにおける治療薬の評価に関連する最善の実践方法について。
ミクログリアとニューロンの相互作用と神経変性疾患
ミクログリアとニューロンの直接的な相互作用、およびこれらの細胞間相互作用が神経変性疾患においてどのように影響を受けるかについて簡潔にレビューします。
ALSマウスモデルを用いた創薬
治療薬の前臨床試験における筋萎縮性側索硬化症(ALS)の研究用動物モデル(マウスおよびラットモデル)の最も効果的な使用方法についてのガイド。
TDP-43 ΔNLS (rNLS8) ALS 治療薬開発用マウス
このリソースは、ALSのΔNLS(deltaNLS、hTDP-43ΔNLS、hTDP-43DeltaNLS、dNLS、TDP43 NLS、rNLS8)TDP-43トランスジェニックマウスモデルの使用に関する情報を提供しており、前臨床治療研究に役立てることができます。
前頭側頭型認知症における神経画像診断と臨床試験
前頭側頭型認知症(FTD)の変異型に対する理解におけるMRIおよびPET画像診断によるバイオマーカーの有用性、およびFTD臨床試験におけるエンドポイントとしてのその使用。