Microglia-Neuron Interactions & Neurodegenerative Diseases
A concise review of the direct interactions between microglia & neurons, and how these cell-to-cell interactions may be affected in neurodegenerative diseases.
Why study direct microglia-neuron interactions?
Microglia are the primary immune cells of the central nervous system and play crucial roles in maintaining brain health and responding to disease. In neurodegenerative diseases, microglia exhibit complex dual functions, with both positive and negative effects on disease progression (Gao, 2023). They help clear misfolded proteins, but they also inadvertently facilitate the spread of these proteins, exacerbating pathology. Microglia provide trophic support and promote an anti-inflammatory environment early in the disease course. However, following sustained reactivity, they can release neurotoxic inflammatory factors.
To better understand and distinguish the complex roles of microglia in neurodegenerative diseases, and how putative therapeutics might influence microglia, it is important to measure microglial phenotypes and their interactions with other cells. While microglia interact both directly and indirectly with most cell types in the brain, this Resource will focus on direct microglia-neuron interactions, which can be readily measured using multiplex immunofluorescence, and how these interactions may be influenced in neurodegenerative diseases.
Where do direct microglia-neuron interactions occur on neurons?
Synapse
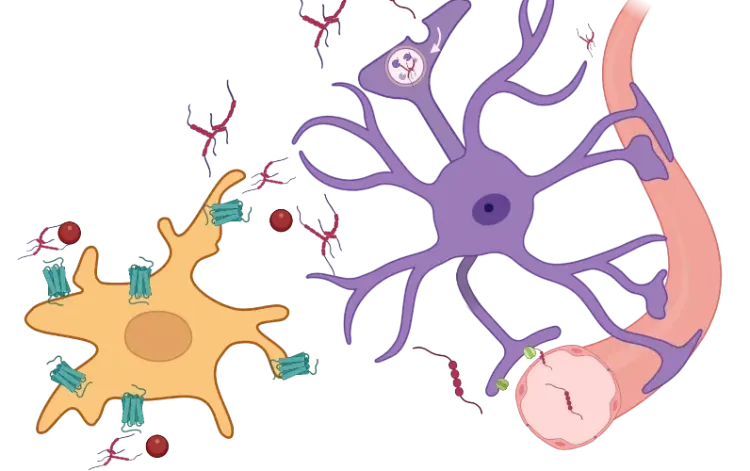
Direct interactions between microglia and neurons at the synapse have been well established. During development, microglia contribute to synaptic pruning (Paolicelli, 2011; Schafer, 2012; Weinhard, 2018) and synapse development via complement signaling (Paolicelli, 2011; Hoshiko, 2012; Zhan, 2014) and fractalkine (CXCL3-CXCR1) signaling (Schafer, 2012). In adulthood, they contribute to the removal of synapses (Trapp, 2007; Yamada, 2008; Tremblay, 2010) and activity-dependent spine remodeling (Parkhurst, 2013). In addition, microglia regulate local network synchrony, post-synaptic current, synaptic scaling, and plasticity (Rogers, 2011; Pfeiffer, 2016; Akiyoshi, 2018; Wang, 2020). Apoptotic synapses can express phosphatidylserine, also known as the “eat-me” signaling molecule, which induces microglial elimination through milk fat globule-EGF factor 8 (MFG-E8) or TAM receptors (Nonaka, 2019). Synapses can be tagged for targeted elimination with C1q, which is recognized by microglial CR3 (complement signaling) (Stevens, 2007; Schafer, 2012).
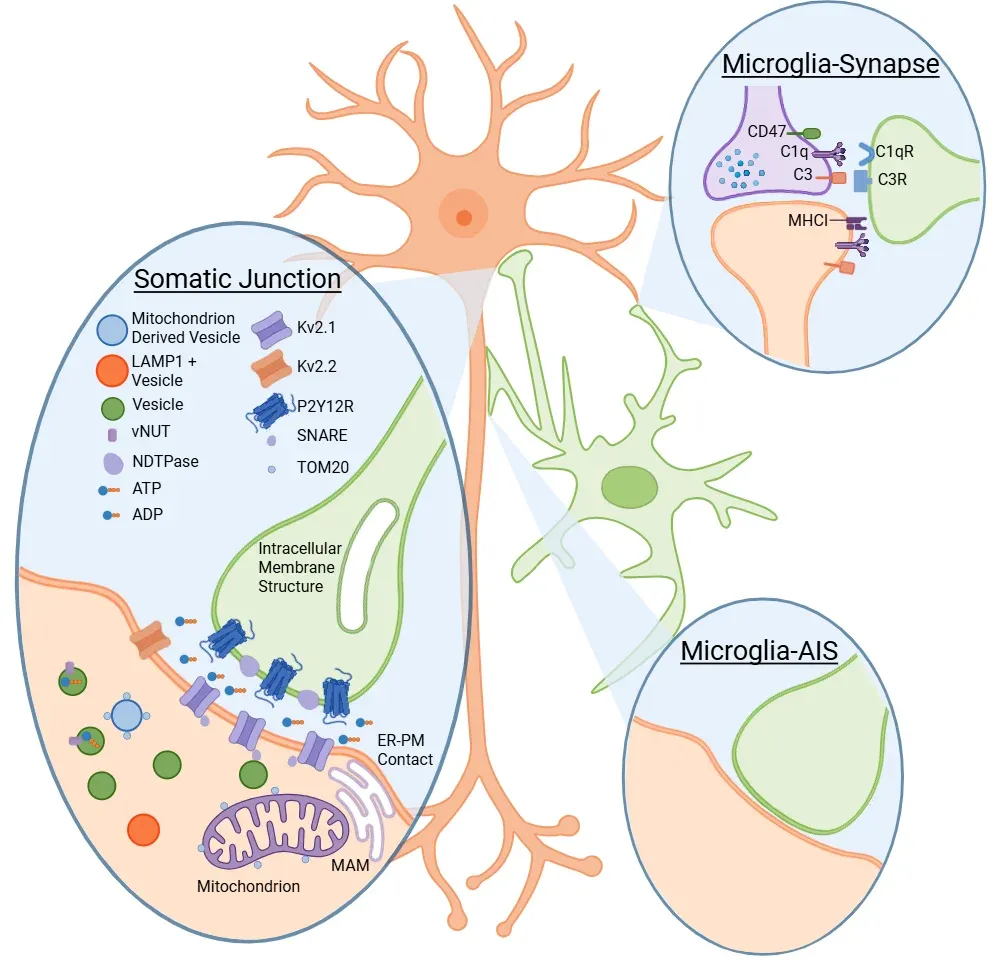
Schematic representation of the direct interactions between microglia and neuronal synapses, soma, and axons, as well as the molecular pathways involved. Adapted from Cserép et al. (Cserép, 2021) under the Creative Commons Attribution License.
Soma
The interactions between microglial processes and neuronal soma are receiving growing research attention (Cserép, 2021). Studies show that microglia can phagocytose neurons during neurodegeneration, acute brain injury, and infection (Sierra, 2010; Fekete, 2018; Janda, 2018). Interestingly, microglia and astrocytes phagocytose different parts of dying neurons. Specifically, microglia migrate and engulf soma and apical dendrites, whereas astrocytes engulf small dendritic apoptotic bodies (Damisah, 2020).
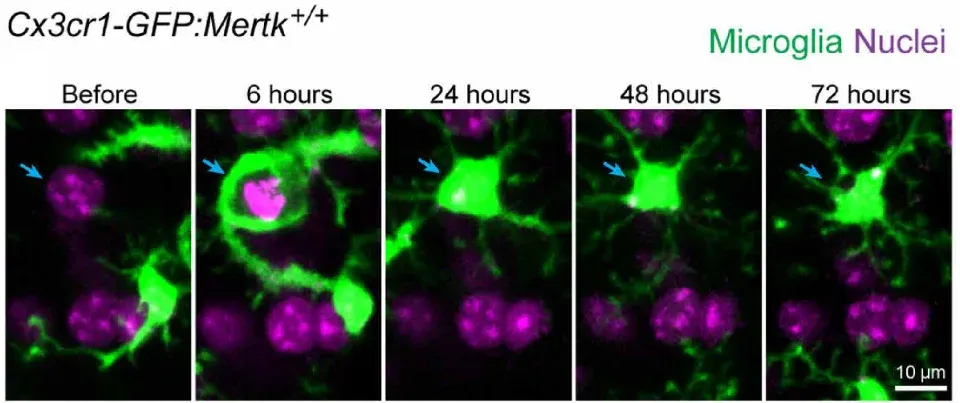
Timelapse imaging of microglial phagocytosis of a dying neuron in a live mouse. Microglia are shown in green (Cx3cr1-GFP) and nuclei (DAPI) in purple. The nucleus of the dying neuron is indicated by a blue arrow. Figure reproduced from Damisah et al. (Damisah, 2020) under the Creative Commons Attribution License.
To specifically phagocytose damaged neurons, microglia must be able to sense neuronal health. Cserép et al. proposed a site to assess neuronal health at a specialized junction found in most neurons, located between microglial processes and neuronal soma, termed the somatic purinergic junction (Cserép, 2020, 2021). On the microglial side, these junctions are enriched in the purinergic receptor P2Y12R, which is necessary for their formation. Whereas, on the neuronal side, the junctions are enriched in Kv2.1 clusters, which are known to play a role in exocytosis, and are associated with mitochondria, mitochondria-associated membrane (MAM), lysosomal-associated membrane protein 1 (LAMP1), and vesicular nucleotide transporter (vNUT). The length of these contacts is three times longer than processes-neurites contacts. They are linked to both neuronal mitochondrial activity and neuronal activity. In an acute brain injury model, studies show an increase in the frequency of these interactions. Further, when these interactions are eliminated through P2Y12R blockage, pathology worsens, suggesting a neuroprotective role of these interactions. Additionally, these somatic junctions are also found in the developing brain, where knockout of P2Y12R results in a disturbed cortical cytoarchitecture (Cserép, 2022).
Neuronal mitochondria, with their diverse roles, not only offer a potential readout of cellular states but also present an opportunity for microglia to influence key neuronal functions. These include firing threshold, proliferation, calcium homeostasis, cell-fate decisions, and metabolism (Hall, 2012; Rugarli, 2012; Chandel, 2014; Kasahara, 2014; Tepikin, 2018; Styr, 2019). While the role of somatic junctions in neurodegenerative diseases remains to be elucidated, mitochondrial function is known to be altered in many neurodegenerative diseases, suggesting that these junctions could be altered in these conditions.
Axon
Microglia also interact with neurons at the axon and the axon initial segment (AIS). While gradients of secreted factors are the main drivers of axonal guidance, microglia also play a role in this process, such as acting as a guidepost (Squarzoni, 2014). In adulthood, microglia migrate toward hyperactive neuronal axons, facilitating membrane repolarization (Kato, 2016). Microglia-mediated clearance of degenerating axons supports neuronal regeneration (Bechmann, 2001; Neumann, 2009), and microglia play a pro-regenerative role in remyelination processes (Lloyd, 2019). Interactions between microglia and the AIS commence early in development, continue throughout the lifespan, and exhibit conservation across species (Baalman, 2015). Moreover, microglia engage diffusely injured axons following traumatic brain injury (Lafrenaye, 2015), and their interactions with the AIS are associated with disrupted AIS integrity in experimental autoimmune encephalomyelitis (Clark, 2016).
How do direct microglia-neuron interactions change in neurodegenerative diseases?
Alzheimer's Disease (AD)
In AD, microglia contribute to neurodegeneration through synaptic engulfment and phagocytosis via complement signaling (C1q, C3) (Hong, 2016; Shi, 2017; Rajendran, 2018; Vogels, 2019) and triggering receptor expressed on myeloid cells 2 (TREM2) signaling (Fracassi, 2023). Interestingly, these direct microglia-neuron interactions appear to be mediated by microglia-astrocyte interactions. Removal of astrocytic APOE4 decreases microglial phagocytosis of synapse- and tau-induced synaptic loss (Wang, 2021). Sokolova et al. (Sokolova, 2024) found a subtype of astrocytes with a unique morphology known as ‘bulbous’ astrocytes that secrete the phagocytic modulator MFG-E8, which is both necessary and sufficient for microglial synapse engulfment in their local environment. Specific astrocytic knockout of Mfge8 reduces synaptic loss and microglia synapse engulfment in two different AD mouse models.
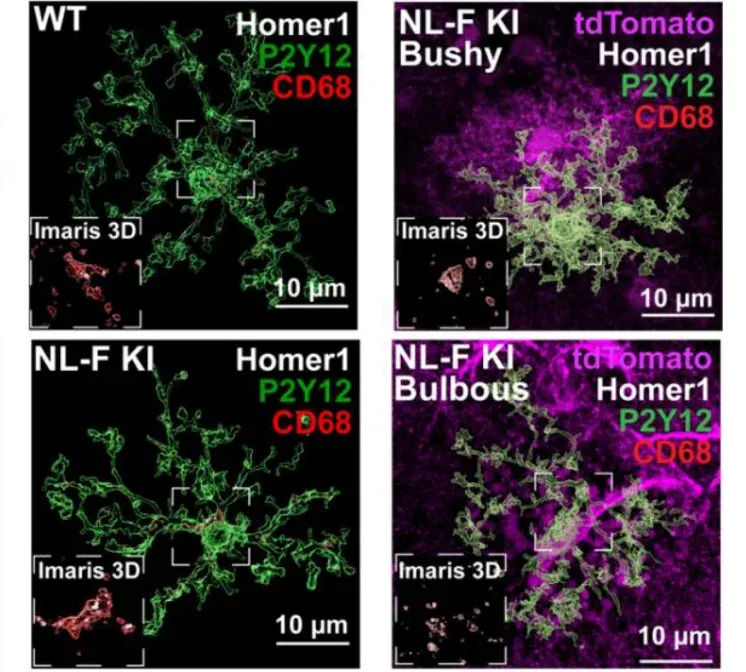
Microglial synapse engulfment in proximity of bulbous astrocytes. Images of excitatory post-synaptic Homer1 (white), CD68 lysosomes (red), astrocytes (purple), and microglia P2Y12 (green). Figure reproduced from Sokolova et al. (Sokolova, 2024) under the Creative Commons Attribution License.
Perineuronal nets (PNNs) are a specialized extracellular matrix (ECM) that surrounds the soma and processes of certain neurons, mostly parvalbumin-expressing (PV+) inhibitory interneurons that play a critical role in cognition (Reichelt, 2019). PNNs play a vital role in synaptic plasticity by stabilizing synapses after the period of heightened plasticity during development. They are also thought to play a protective role by acting as a physical barrier to neurons they surround. Crapser et al. (Crapser, 2020) showed that PNNs decrease in a AD mouse model as well in human AD brains. This decrease in PNNs is followed by loss of PV+ interneurons. Microglia engulf PNNs material and depletion of microglia prevents PNNs loss. As PNNs have been reported to protect neurons against tau and amyloid aggregates (Brückner, 1999; Miyata, 2007; Suttkus, 2016; Reichelt, 2019), this microglia-neuron interaction could be another axis for neurodegeneration.
von Saucken et al. (von Saucken, 2020) found that prior to plaque accumulation, microglia display morphological changes, and they preferentially engage neurons containing amyloid-beta. These direct microglial-neuronal contacts are measured from processes to soma, soma to soma, and processes to neurites. Microglial phagocytosis of neurites is higher in regions with more amyloid-beta burden. While TREM2 is necessary for morphological changes, it is not necessary for the preferential engagement of amyloid-bearing neurons or the difference in neurite phagocytosis. Interestingly, in an MPTP mouse model of Parkinson’s Disease (PD), an increase in soma-soma contact occurs prior to microglial engulfment and phagocytosis of neurons (Barcia, 2012), suggesting that this process could be part of the amyloid-beta model.
Puigdellívol et al. found a role for microglial phagocytosis of neurons in both amyloid-beta injection and P301S tau mouse models of AD (Puigdellívol, 2021). Knockout of the microglial P2Y6 receptor (P2Y6R), which is required for microglial phagocytosis of neurons, prevents both neuronal loss and memory loss.
Tauopathies
Similarly, microglia engulf synapses in the tau-P301S transgenic AD mouse model leading to a decline in synaptic density (Dejanovic, 2018). C1q-tagging correlates with phospho-tau accumulation in the postsynaptic densities (PSD), and a C1q-blocking antibody reduces microglial synapse engulfment and increases synaptic density.
In addition to extracellular tau, microglia phagocytose live neurons containing tau aggregates (Brelstaff, 2018). Secretion of MFG-E8 by microglia is necessary for the phagocytosis of aggregates-bearing neurons in cell co-cultures, but not for the localization of microglia to these neurons in a mouse model of tauopathy. Moreover, in human brain extracts from both FTDP-17 and Pick’s frontotemporal dementia (FTD) patients there is increased levels of MFG-E8, but not in C9orf72 FTD patients.
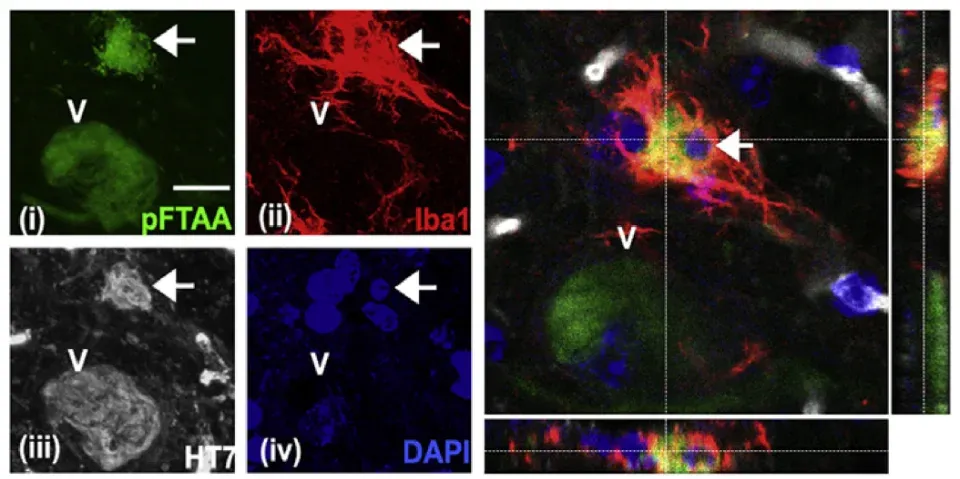
Engulfment of a neuron containing tau aggregates (pFTAA, green) by microglia (Iba-1, red) in 5 month-old P301S mice, with human (HT7) P301S-tau shown in white and nuclei (DAPI, blue). A confocal z-stack shows complete engulfment of the neuron (indicated by a white arrow) by the microglia. Figure reproduced from Brelstaff et al. (Brelstaff, 2018) under the Creative Commons Attribution License.
The direct interaction between neuronal membrane-bound CX3CL1 and microglia CX3CR1 is thought to reduce inflammation and keep microglia in a surveillant state (Szepesi, 2018). Accordingly, knockouts of CX3CL1 (Lee, 2014; Bemiller, 2018) or CX3CR1 (Bhaskar, 2010) enhances tau phosphorylation and neuroinflammation in mouse models. The cleaved, soluble ligand CX3CL1 does not rescue the knockouts (Lee, 2014; Bemiller, 2018), indicating that these interactions happen in a juxtacrine manner. It is currently unknown on which compartment(s) these interactions occur. However, the somatic purinergic junctions could present an interesting site, as the lifetime of these interactions has been shown to be more than three times longer than on neurites, which would provide more opportunities for signaling to occur.
Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD)
TDP-43 aggregates are a hallmark of ALS, and about half of patients with FTD have TDP-43 aggregates. In ALS, changes in gene expression in microglia occur before neurons (Maniatis, 2019). Microglia also become reactive, and microgliosis correlates with patients upper motor neurons clinical scores (Brettschneider, 2012). Using the inducible TDP-43ΔNLS mouse model, Xie et al. (Xie, 2024) show an increase in neuronal cortical hyperactivity early in the disease course. This cortical hyperactivity is followed by the appearance of rod-shaped microglia, peaking at 3 weeks off dox and declining after. The rod-shaped microglia interact closely with dendrites, particularly the apical dendrites of pyramidal neurons, aligning along their axes and extensively phagocytosing excitatory synapses. Knockout of TREM2 reduces the fraction of non-ramified (including rod-shaped) microglia, increasing the neuronal hyperactivity and the motor deficits, and reducing survival.
Models of familial FTD and ALS also shed light on microglia-neuron interactions. Progranulin deficiency through GRN mutations is one of the main causes of genetic FTD. Grn-/- mice exhibit an age-dependent TDP-43 proteinopathy as well as microglial reactivity (Lui, 2016; Zhang, 2020). This model also undergoes microglial-dependent synaptic pruning and neurodegeneration, which are rescued by C1q and/or C3 deletion (Lui, 2016; Zhang, 2020).
Repeat expansions in C9orf72 are a genetic cause of FTD and ALS. C9orf72 deficiency in mice shifts microglia away from the homeostatic static to an inflammatory state (Lall, 2021). C9orf72-/- mice have age-dependent cortical synaptic pruning and deficiency in learning and memory behaviors. The enhanced synaptic pruning is recapitulated by knocking out C9orf72 specifically in microglia. In addition, an AD mouse model with C9orf72 deficiency (5XFAD/C9orf72-/-) also shows enhanced synaptic pruning, but with a reduced plaque load.
Our team would be happy to answer any questions about microglia-neuron interactions & neurodegenerative diseases or provide specific information about the neurodegenerative disease models we use for therapeutic efficacy studies.
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on Neuroinflammation and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
Microglia Morphology in ALS, Alzheimer's Disease & Parkinson's Disease
An overview of microglial morphological analysis and the applications to neurodegenerative disease research and drug discovery & development.
Microglia, Astrocytes & Tau in Neurodegenerative Diseases
How glial-driven neuroinflammation fuels tau aggregation, propagation, and neuronal loss in Alzheimer’s disease and other tauopathies.
Mitochondrial Dysfunction in Microglia & Astrocytes
The role of mitochondrial dysfunction in microglia and astrocytes in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and ALS.
Microglial Activation in an α-Synuclein PFF Mouse Model
We have quantified microglial activation, based on morphology, in an α-synuclein preformed fibril (PFF) seeding & spreading mouse model of Parkinson’s disease.
Amyloid-β & Inflammatory Microenvironment in Alzheimer's Mice
We have analyzed the complex spatial relationships between β-amyloid plaques, activated & resting microglia, and astrocytes in an APP/PS1 transgenic model.