TDP-43: Role in ALS and Frontotemporal Dementia (FTD)
TDP-43 (TAR DNA-binding protein 43) is a DNA- and RNA-binding protein that plays a central role in the pathology of amyotrophic lateral sclerosis (ALS) & frontotemporal dementia (FTD). In these neurodegenerative diseases, TDP-43 abnormally mislocalizes and aggregates in neurons and glial cells, disrupting RNA processing and contributing to progressive neuronal dysfunction. Understanding TDP-43 pathology is critical for advancing disease models, biomarkers, and therapeutic development in ALS and FTD.
What is TDP-43?
TDP-43 (sometimes written as TPD43) is a DNA- and RNA-binding protein that abnormally accumulates in neurons and glial cells in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), where it disrupts RNA processing and leads to neurodegeneration.
TAR DNA Binding Protein (TDP-43) is a highly conserved protein that binds RNA and DNA. It is encoded in the TARDBP gene and belongs to the heterogeneous nuclear ribonucleoprotein (hnRNP) family within the nucleus (Cohen, 2011; de Boer, 2020; Jo, 2020; Corbet, 2021; Tziortzouda, 2021).
Under normal physiological conditions, TDP-43 is primarily found in the nucleus, but it also shuttles actively between the nucleus and the cytoplasm. When mislocalized to the cytoplasm, TDP-43 can lead to proteinopathies such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). In these pathologies, it forms harmful aggregates that contribute to neurodegeneration (de Boer, 2020; Jo, 2020; Chhangani, 2021; Scialo, 2025).
TDP-43 Functions
TDP-43 plays a crucial role in regulating gene expression and RNA processing within the nucleus.
Nuclear functions of TDP-43 include (de Boer, 2020; Corbet, 2021; Dykstra, 2025):
- RNA transcriptional regulation by repressing the inclusion of cryptic exons during splicing
- Maintenance, metabolism, and transport of mRNA
- Maturation of microRNAs (miRNAs)
TDP-43 moves to the cytoplasm to perform different functions, and it is regulated through a negative feedback mechanism.
Cytoplasmic functions of TDP-43 include (Cohen, 2011; Jo, 2020; Corbet, 2021; Tziortzouda, 2021):
- Stability and translation of mRNA
- Formation of two structures:
- Stress granules, which are essential for neuronal protection against oxidative stress
- Ribonucleoprotein granules, important for mRNA transport and miRNA biogenesis
In addition, TDP-43 associates with the mitochondrial genome and is involved in pathways related to the respiratory chain. This protein is critical for the normal development of central neuronal cells during the early stages of embryogenesis. In fact, in mouse models, a loss of TDP-43 function has been shown to be embryonically lethal (Cohen, 2011; de Boer, 2020).
The diverse functions of TDP-43 originate from its various regions (Cohen, 2011; de Boer, 2020; Jo, 2020; Tziortzouda, 2021; Corbet, 2021):
- The N-terminal domain (NTD) promotes self-oligomerization of TDP-43. This domain also includes the nuclear localization signal (NLS), which is important for transporting the protein into the nucleus.
- The RNA recognition motifs (RRM1 and RRM2) are responsible for binding to nucleic acids, including both RNA and DNA.
- The nuclear export signal (NES) facilitates the transport of TDP-43.
- The C-terminal domain (CTD) regulates protein solubility and mediates pathological aggregation, as well as contributing to recruiting TDP-43 into stress granules.
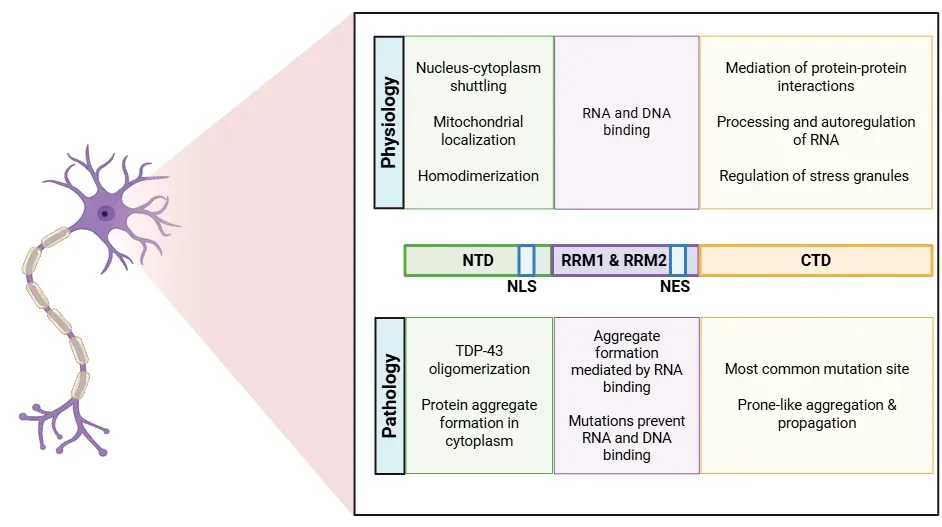
Structure of the TDP-43 protein, detailing physiological and pathological roles of each site.
What is the significance of TDP-43 pathology in ALS and FTD?
TDP-43 proteinopathy is a hallmark of several neurodegenerative diseases, including ALS and FTD. The aggregation of TDP-43 protein is closely linked to disease progression and severity. TDP-43 aggregates can also be found in 20-50% of Alzheimer’s disease (AD) patients, where they correlate with poorer memory performance and increased brain atrophy (Prasad, 2019).
The pathogenic process associated with TDP-43 proteinopathy includes both toxic gain-of-function and loss-of-function mechanisms. TDP-43 cleavage, hyperphosphorylation, and ubiquitination lead to the formation of toxic aggregates in both the nucleus and cytoplasm (Mackenzie, 2008; Xu, 2014; Yang, 2014; Scotter, 2015; Meneses, 2021).
Various gene mutations have been described in TDP-43 pathology, with many of these localized in the CTD. For instance, TDP-43 pathology may arise from a loss of nuclear TDP-43 due to mis-splicing of genes, resulting in the inclusion of cryptic exons. Two relevant genes affected by these mutations are:
- UNC13A: The loss of functional TDP-43 results in the inclusion of a cryptic exon in UNC13A mRNA, which leads to a decrease in the expression of the UNC13A protein, important for synaptic transmission (Garcia-Montojo, 2024).
- Stathmin2 (STMN2): STMN2 is important for motor neuron growth and repair. Loss of TDP-43 function leads to mis-splicing and depletion of STMN2 due to the inclusion of a cryptic exon and polyadenylation (Suk, 2020; Ma, 2022).
Other significant mutations linked to TDP-43 pathology involve the C9orf72 gene (which is associated with classic ALS and FTD), TARDBP, and ALS2 genes (de Boer, 2020; Jo, 2020).
Under pathological circumstances, TDP-43 is increasingly transported from the nucleus to the cytoplasm through several mechanisms (Yang, 2014; Prasad, 2019; Suk, 2020; Garcia-Montojo, 2024):
- A mutation in the NLS within NTD.
- Disruption of stress granule regulation: TDP-43, RNA, and other proteins accumulate in the cytoplasm following oxidative stress or heat shock. In physiological conditions, TDP-43 typically relocates back to the nucleus once the cellular stress dissipates.
- Disruption of the nuclear pore complex: The reduction of (G4C2)30 RNA levels decreases the expression of Ran, which regulates the nuclear localization of TDP-43. Other proteins, such as Nup62 and Kpnb1, also contribute to this transport process.
- Cytoplasmic aggregates: The formation of cytoplasmic aggregates of TDP-43 leads to a vicious cycle that further disrupts nucleocytoplasmic transport and the nuclear pore complexes, causing even more TDP-43 to exit the nucleus.
These pathological changes result in:
- Loss of function (Yang, 2014; de Boer, 2020; Suk, 2020; Garcia-Montojo, 2024):
- Dysregulation of RNA metabolism
- Depletion of nuclear TDP-43
- Impaired autophagy
- Mitochondrial dysfunction
- Toxic gain of function (Xu, 2014; Prasad, 2019; Suk, 2020; Meneses, 2021):
- Formation of pathological polymers leading to increased protein aggregation and neurotoxicity
- Prion-like propagation via exosomes or synaptic transmission. Microglia, astrocytes, and oligodendrocytes can phagocyte TDP-43 and transmit it to neurons
- See our Resource: Microglia-Neuron Interactions & Neurodegenerative Diseases
- Ultimately, the presence of TDP-43 aggregates contributes to neuronal death
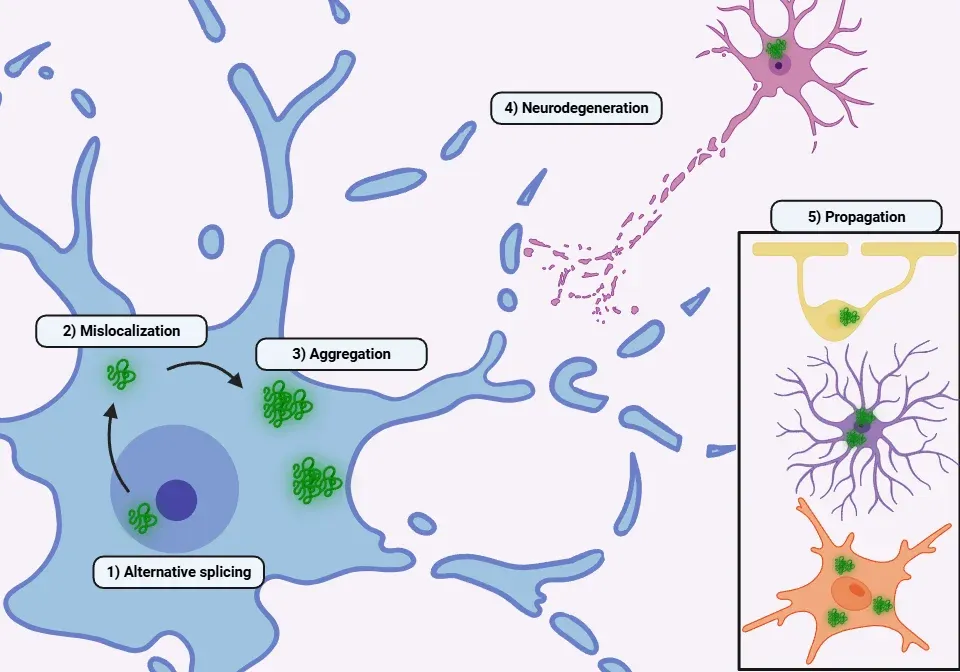
An overview of the pathophysiological mechanisms underlying TDP-43 proteinopathies.
ALS
Cytoplasmic aggregation of TDP-43 is considered the main pathological hallmark of ALS (Xu, 2014; Scotter, 2015; Prasad, 2019; de Boer, 2020; Suk, 2020; Hu, 2024):
- Over 90% of patients with sporadic and familial ALS exhibit TDP-43 aggregates.
- While ALS caused by mutations in SOD1 or FUS typically does not show TDP-43 pathology, mutations in TARDBP lead to TDP-43 pathology and are a direct cause of familial ALS.
- However, TARDBP mutations are found in only about 4% of familial ALS cases, which accounts for less than 10% of all ALS cases.
- Other genetic mutations associated with TDP-43 aggregation in ALS include A90V (located in the NLS region), G294V, and A315T.
- Symptoms such as executive dysfunction, language difficulties, and behavioral abnormalities can occur depending on the location of the TDP-43 pathology.
Three types of TDP-43 pathology have been identified in ALS (de Boer, 2020):
- Mixed neuronal and glial pathology
- Glial pathology (particularly in astrocytes)
- Neuronal pathology
TDP-43 pathology is most prominent in cortical layer V neurons and spinal motor neurons. Tissues from ALS patients show regional variations in the species of TDP-43 deposited (Scotter, 2015; Wu, 2024):
- C-terminal truncated fragments are enriched in the brain
- Full-length TDP-43 is predominant in spinal cord inclusions
A partial loss of TDP-43 function in mouse models is sufficient to cause neurodegeneration, progressive motor dysfunction, paralysis, and ultimately death (Xu, 2014; Yang, 2014).
See: TDP-43 Mouse Model of Amyotrophic Lateral Sclerosis (ALS)
FTD
FTD, also known as frontotemporal lobar degeneration (FTLD) from a pathological perspective, is characterized by the presence of TDP-43, tau or FUS protein aggregates in neurons and glial cells (Prasad, 2019; Ho, 2024; Hu, 2024).
- TDP-43 mutations are associated with only a small percentage of FTD cases.
- For instance, the C9orf72 mutation accounts for approximately 13% of FTD cases.
- However, TDP-43 aggregates are found in up to 50% of FTD patients.
- Another important mutation is the GRN gene (Progranulin), which leads to loss of function and is inherited in an autosomal dominant manner.
Symptoms of FTD related to TDP-43 include changes in personality, behavior, and language skills, such as loss of empathy, inappropriate social behavior, and difficulty communicating (Prasad, 2019; de Boer, 2020; Jo, 2020).
See our Resource: Neuroimaging in Frontotemporal Dementia & Clinical Trials
In patients with FTD, total serum TDP-43 levels are often significantly lower than those in healthy controls, particularly among carriers of the C9orf72 repeat expansion or those with FTD with concomitant motor neuron disease phenotype. This decrease in levels is hypothesized to result from the protein being sequestered into insoluble aggregates (Katisko, 2022).
There are three different types of pathology reported in FTLD: FTLD-tau (35-50%), FTLD-FUS (10%), and FTLD-TDP (approximately 50%). FTLD-TDP is further divided into the following subtypes (Prasad, 2019; de Boer, 2020; Meneses, 2021):
- Type A: Compact or crescent-shaped neuronal cytoplasmic inclusions (NCI), usually found in the neocortex.
- Type B: Diffuse or granular NCIs, often located in cortical and subcortical oligodendrocytes, and associated with motor neuron disease.
- Type C: Numerous dystrophic neurites, predominantly in the neocortex.
- Type D: Numerous neuronal intranuclear inclusions.
See our Innovations: Diffusion MRI & Frontotemporal Dementia (FTD) & Frontotemporal Dementia (FTD) & MRI Brain Atrophy
Is TDP-43 a potential therapeutic target?
Current therapeutic strategies emphasize three main approaches: removing the toxic protein, restoring its function, and addressing the downstream effects of its depletion.
Vectorized Antibodies
- ACI-5891 is a vectorized monoclonal antibody delivered using AAV9, specifically targeting pathological TDP-43. In mouse models of ALS and FTD, a single intracisternal injection successfully reduced pathological phospho-TDP-43 signals by up to 68% (Val, 2025).
- The antibody 3B12A detects the abnormal exposure of residues E246 and D247 in misfolded TDP-43 aggregates and is engineered to promote proteasome degradation of TDP-43 (Francois-Moutal, 2021).
- VH7Vk9 promotes proteasome or autophagy-driven degradation by focusing on the RRM1 domain (Francois-Moutal, 2021).
Small Molecules
- Baicalein acts as a structure-correcting agent, helping to transform existing misfolded proteins into functional forms (Chang, 2024).
- M102 is an activator of the Nuclear Factor Erythroid 2-Related Factor 2 (NRF2) and Heat Shock Factor 1 (HSF-1) pathways, showing promise in animal models of ALS by reducing oxidative stress and the effects of TDP-43 aggregation (San Gil, 2025).
- Additional molecules have been explored in fly models (Francois-Moutal, 2021):
- rTRD0, which targets the RRM1 nucleic acid binding interface
- nTRD22, which targets the N-terminal domain to promote TDP-43 clearance
Antisense Oligonucleotides (ASO)
- QRL-201 targets STMN2, a protein mis-spliced in TDP-43 pathology, which helps improve motor neuron function. This therapy is currently in Phase 1 trials for sporadic and C9orf72-ALS (Liu, 2024).
- Qalsody is FDA-approved for patients with ALS featuring a SOD1 mutation, but does not address TDP-43 pathology (San Gil, 2025).
Gene Therapies
- Under preclinical development to correct the aberrant splicing of other affected genes, such as UNC13a (San Gil, 2025).
Chaperone-Targeting Therapies
- Overexpressing J-domain proteins (DNAJB1, DNAJB2a, DNAJB4, DNAJB5) significantly reduces insoluble TDP-43 in cell models (San Gil, 2025).
14-3-3θ
- Targets misfolded TDP-43 in mouse models of ALS, leading to improvements in behavioral deficits (San Gil, 2025).
Autophagy Induction
- Improves the clearance of protein aggregates (Hayes, 2022):
- mTOR inhibitors like Rapamycin
- TFEB inducers such as Trehalose and Ibudilast
- See our Resources: Impaired Microglia Autophagy in Neurodegenerative Diseases & Autophagy and Transcription Factor EB (TFEB)
The C-terminal glycine-rich domain and the RRM2 domain are crucial in driving phase separation and aggregation, making them attractive targets for specific antibodies (Riemenschneider, 2023). As the authors stated, caution is necessary when targeting essential domains, as active immunization with some N-terminal peptides caused lethality in mice, likely due to the physiological role of TDP-43 in the nucleus (Riemenschneider, 2023).
Several research groups have demonstrated disease modification through therapeutic interventions in TDP-43 ΔNLS ALS mouse models. Notable findings include:
- AIT-101: A small molecule that inhibits PIKfyve. This treatment has been shown to reduce body weight loss, improve motor deficits, and lower levels of neurofilament light (NfL) in both plasma and CSF, as well as decrease TDP-43 aggregates (Young, 2023).
- VRG50304: This compound has been found to reduce plasma and CSF NfL levels, as well as decrease acid sphingomyelinase activity in the causal cortex (Stomakhina, 2021).
- AAV9/NF242: Intracerebroventricular injections of the N-terminal fragment of rho guanine nucleotide exchange factor have improved both the lifespan and motor performance of the mice (Droppelmann, 2024).
See our Resource for more details: TDP-43 ΔNLS (rNLS8) Mice for ALS Drug Development
While several therapies targeting TDP-43 have failed in ALS clinical trials, such as arimoclomol, sodium phenylbutyrate and ursodoxicoltaurine, colchicine, and BIIB105 (San Gil, 2025), TDP-43 continues to be an important target in ongoing research. Although no TDP-43-based therapies have yet successfully demonstrated the ability to slow disease progression in humans, many are in the preclinical stages of development, providing a strong foundation for future advancements. Progressing towards effective treatments will need innovative strategies that address both TDP-43 aggregation and its functional loss, paving the way for potential breakthroughs in ALS therapy (San Gil, 2025).
Our team would be happy to answer any questions about TDP-43 or provide specific information about the models that we use for therapeutic efficacy studies.
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on Multiple Sclerosis and best practices related to the evaluation of therapeutic agents in MS animal models.
Microglia-Neuron Interactions & Neurodegenerative Diseases
A concise review of the direct interactions between microglia & neurons, and how these cell-to-cell interactions may be affected in neurodegenerative diseases.
A Guide to ALS Models for Drug Discovery
A Resource for the most effective use of research animal models (mouse & rat models) of Amyotrophic Lateral Sclerosis (ALS) for preclinical testing of therapeutics.
TDP-43 ΔNLS (rNLS8) Mice for ALS Drug Development
This resource provides information about the use of the ΔNLS (deltaNLS, hTDP-43ΔNLS, hTDP-43DeltaNLS, dNLS, TDP43 NLS, rNLS8) TDP-43 transgenic mouse model of ALS for preclinical therapeutic studies.
Neuroimaging in Frontotemporal Dementia & Clinical Trials
The utility of MRI & PET imaging biomarkers in our understanding of Frontotemporal Dementia (FTD) variants, and their use as endpoints in FTD clinical trials.
Diffusion MRI & Frontotemporal Dementia (FTD)
Diffusion neuroimaging analysis from the FTLDNI natural history study of Frontotemporal Dementia (FTD).
Frontotemporal Dementia (FTD) & MRI Brain Atrophy
Neuroimaging biomarkers (including MRI brain atrophy) from the FTLDNI natural history study of Frontotemporal Dementia (FTD).