TDP-43 - Son rôle dans la SLA et la DFT
Présentation générale de la TDP-43, de son rôle physiologique, de son importance dans la pathologie de la SLA et de la DFT, ainsi que des stratégies thérapeutiques impliquant la TDP-43.
Qu'est-ce que la TDP-43?
La protéine de liaison à l'ADN à réponse transactive (TDP-43) est une protéine hautement conservée qui se lie à l'ARN et à l'ADN. Elle est codée par le gène TARDBP et appartient à la famille des ribonucléoprotéines nucléaires hétérogènes (hnRNP) présentes dans le noyau (Cohen, 2011; de Boer, 2020; Jo, 2020; Corbet, 2021; Tziortzouda, 2021).
Dans des conditions physiologiques normales, la TDP-43 se trouve principalement dans le noyau, mais elle fait également la navette entre le noyau et le cytoplasme. Lorsqu'elle est mal localisée dans le cytoplasme, la TDP-43 peut entraîner des protéinopathies telles que la sclérose latérale amyotrophique (SLA) et la démence frontotemporale (DFT). Dans ces pathologies, elle forme des agrégats nocifs qui contribuent à la neurodégénérescence (de Boer, 2020; Jo, 2020; Chhangani, 2021; Scialo, 2025).
Fonctions de la TDP-43
La TDP-43 joue un rôle crucial dans la régulation de l'expression génique et le traitement de l'ARN au sein du noyau.
Les fonctions nucléaires de la TDP-43 comprennent (de Boer, 2020; Corbet, 2021; Dykstra, 2025):
- La régulation transcriptionnelle de l'ARN en réprimant l'inclusion d'exons cryptiques pendant l'épissage
- Le maintien, le métabolisme et le transport de l'ARNm
- La maturation des microARN (miARN)
La TDP-43 se déplace vers le cytoplasme pour remplir différentes fonctions, et elle est régulée par un mécanisme de rétroaction négative.
Les fonctions cytoplasmiques de la TDP-43 comprennent (Cohen, 2011; Jo, 2020; Corbet, 2021; Tziortzouda, 2021):
- Stabilité et traduction de l'ARNm
- Formation de deux structures :
- Les granules de stress, qui sont essentiels à la protection des neurones contre le stress oxydatif
- Les granules ribonucléoprotéiques, importants pour le transport de l'ARNm et la biogenèse des miARN
De plus, la TDP-43 s'associe au génome mitochondrial et intervient dans les voies liées à la chaîne respiratoire. Cette protéine est essentielle au développement normal des cellules neuronales centrales au cours des premiers stades de l'embryogenèse. En effet, chez les modèles murins, la perte de la fonction TDP-43 s'est avérée létale pour l'embryon (Cohen, 2011; de Boer, 2020).
Les diverses fonctions de la TDP-43 proviennent de ses différentes régions (Cohen, 2011; de Boer, 2020; Jo, 2020; Tziortzouda, 2021; Corbet, 2021):
- Le domaine N-terminal (NTD) favorise l'auto-oligomérisation de la TDP-43. Ce domaine comprend également le signal de localisation nucléaire (NLS), qui est important pour le transport de la protéine vers le noyau.
- Les motifs de reconnaissance de l'ARN (RRM1 et RRM2) sont responsables de la liaison aux acides nucléiques, y compris l'ARN et l'ADN.
- Le signal d'export nucléaire (NES) facilite le transport de la TDP-43.
- Le domaine C-terminal (CTD) régule la solubilité des protéines et intervient dans l'agrégation pathologique, tout en contribuant au recrutement de la TDP-43 dans les granules de stress.
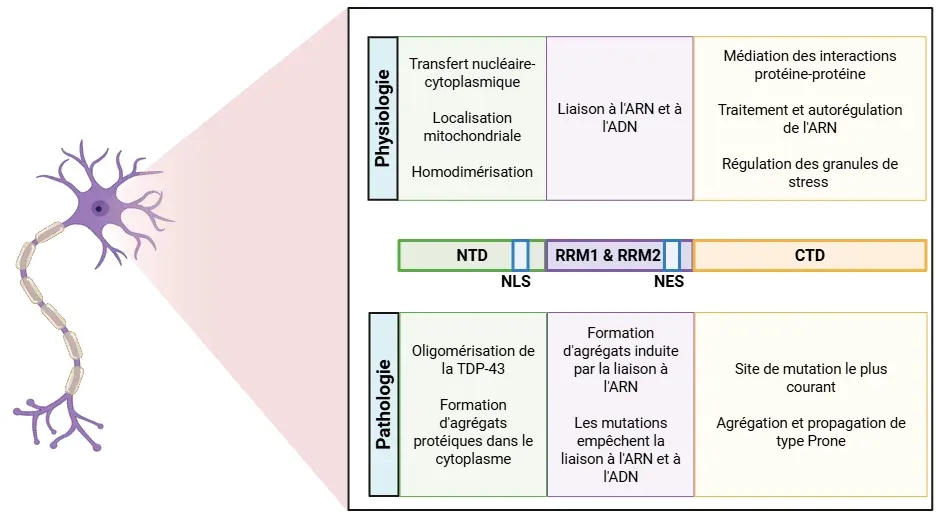
Structure de la protéine TDP-43, détaillant les rôles physiologiques et pathologiques de chaque site.
Quelle est l'importance de la pathologie TDP-43 dans la SLA et la DFT?
La protéinopathie TDP-43 est caractéristique de plusieurs maladies neurodégénératives, notamment la SLA et la DFT. L'agrégation de la protéine TDP-43 est étroitement liée à la progression et à la gravité de la maladie. On trouve également des agrégats de TDP-43 chez 20 à 50 % des patients atteints de la maladie d'Alzheimer (MA), où ils sont associés à une détérioration de la mémoire et à une atrophie cérébrale accrue (Prasad, 2019).
Le processus pathogène associé à la protéinopathie TDP-43 comprend à la fois des mécanismes toxiques de gain de fonction et de perte de fonction. Le clivage, l'hyperphosphorylation et l'ubiquitination de la TDP-43 conduisent à la formation d'agrégats toxiques dans le noyau et le cytoplasme (Mackenzie, 2008; Xu, 2014; Yang, 2014; Scotter, 2015; Meneses, 2021).
Diverses mutations génétiques ont été décrites dans la pathologie TDP-43, dont beaucoup sont localisées dans le CTD. Par exemple, la pathologie TDP-43 peut résulter d'une perte de TDP-43 nucléaire due à un mauvais épissage des gènes, entraînant l'inclusion d'exons cryptiques. Deux gènes pertinents affectés par ces mutations sont :
- UNC13A: la perte de la TDP-43 fonctionnelle entraîne l'inclusion d'un exon cryptique dans l'ARNm de l'UNC13A, ce qui conduit à une diminution de l'expression de la protéine UNC13A, importante pour la transmission synaptique (Garcia-Montojo, 2024).
- Stathmin2 (STMN2): le gène STMN2 est important pour la croissance et la réparation des motoneurones. La perte de la fonction TDP-43 entraîne un épissage incorrect et une déplétion du gène STMN2 en raison de l'inclusion d'un exon cryptique et d'une polyadénylation (Suk, 2020; Ma, 2022).
D'autres mutations importantes liées à la pathologie TDP-43 concernent les gènes C9orf72 (associé à la SLA classique et à la DFT), TARDBP et ALS2 (de Boer, 2020; Jo, 2020).
Dans des circonstances pathologiques, la TDP-43 est de plus en plus transportée du noyau vers le cytoplasme par plusieurs mécanismes (Yang, 2014; Prasad, 2019; Suk, 2020; Garcia-Montojo, 2024):
- Une mutation dans le NLS au sein du NTD.
- Perturbation de la régulation des granules de stress : la TDP-43, l'ARN et d'autres protéines s'accumulent dans le cytoplasme à la suite d'un stress oxydatif ou d'un choc thermique. Dans des conditions physiologiques, la TDP-43 retourne généralement dans le noyau une fois que le stress cellulaire s'est dissipé.
- Perturbation du complexe des pores nucléaires : la réduction des niveaux d'ARN (G4C2)30 diminue l'expression de Ran, qui régule la localisation nucléaire de la TDP-43. D'autres protéines, telles que Nup62 et Kpnb1, contribuent également à ce processus de transport.
- Agrégats cytoplasmiques : la formation d'agrégats cytoplasmiques de TDP-43 entraîne un cercle vicieux qui perturbe davantage le transport nucléocytoplasmique et les complexes des pores nucléaires, provoquant la sortie d'une quantité encore plus importante de TDP-43 du noyau.
Ces changements pathologiques entraînent :
- Perte de fonction (Yang, 2014; de Boer, 2020; Suk, 2020; Garcia-Montojo, 2024):
- Une dérégulation du métabolisme de l'ARN
- Dépérissement de la TDP-43 nucléaire
- Altération de l'autophagie
- Dysfonctionnement mitochondrial
- Gain de fonction toxique (Xu, 2014; Prasad, 2019; Suk, 2020; Meneses, 2021) :
- Formation de polymères pathologiques entraînant une augmentation de l'agrégation des protéines et de la neurotoxicité
- Propagation de type prion via les exosomes ou la transmission synaptique. Les microglies, les astrocytes et les oligodendrocytes peuvent phagocyter la TDP-43 et la transmettre aux neurones
- Veuillez consulter notre ressource : Interactions entre les microglies et les neurones et maladies neurodégénératives
- En fin de compte, la présence d'agrégats de TDP-43 contribue à la mort neuronale
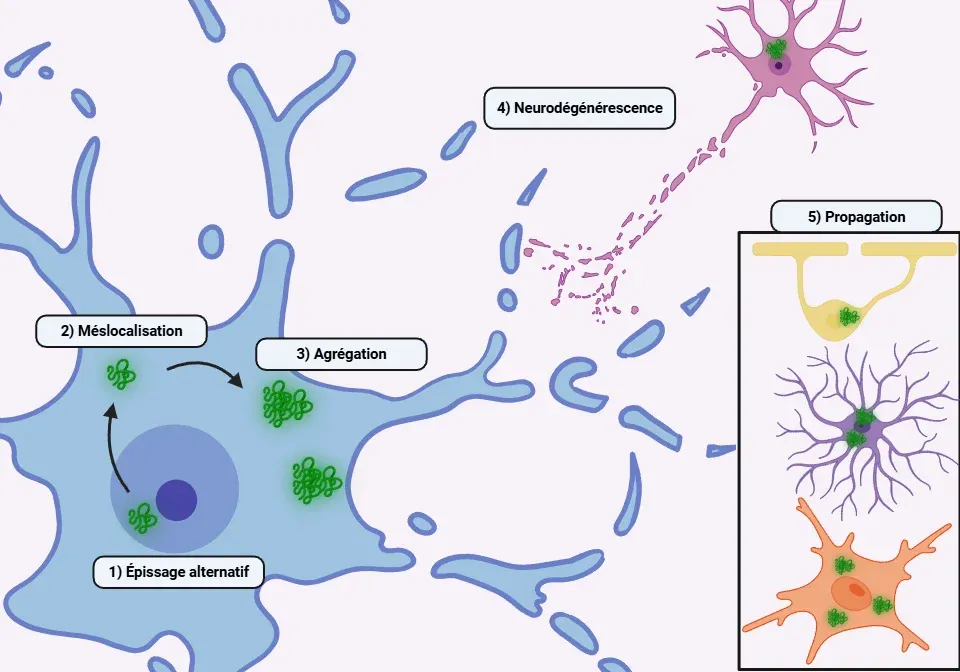
Présentation des mécanismes physiopathologiques sous-jacents aux protéinopathies TDP-43.
SLA
L'agrégation cytoplasmique de la TDP-43 est considérée comme la principale caractéristique pathologique de la SLA (Xu, 2014; Scotter, 2015; Prasad, 2019; de Boer, 2020; Suk, 2020; Hu, 2024):
- Plus de 90 % des patients atteints de SLA sporadique et familiale présentent des agrégats de TDP-43.
- Alors que la SLA causée par des mutations dans les gènes SOD1 ou FUS ne présente généralement pas de pathologie TDP-43, les mutations dans le gène TARDBP conduisent à une pathologie TDP-43 et sont une cause directe de la SLA familiale.
- Cependant, les mutations du gène TARDBP ne sont observées que dans environ 4 % des cas de SLA familiale, ce qui représente moins de 10 % de tous les cas de SLA.
- D'autres mutations génétiques associées à l'agrégation de la TDP-43 dans la SLA comprennent A90V (située dans la région NLS), G294V et A315T.
- Des symptômes tels que des troubles exécutifs, des difficultés linguistiques et des anomalies comportementales peuvent apparaître en fonction de la localisation de la pathologie TDP-43.
Trois types de pathologie TDP-43 ont été identifiés dans la SLA (de Boer, 2020):
- Pathologie neuronale et gliale mixte
- Pathologie gliale (en particulier dans les astrocytes)
- Pathologie neuronale
La pathologie TDP-43 est plus marquée dans les neurones de la couche corticale V et les motoneurones spinaux. Les tissus des patients atteints de SLA présentent des variations régionales dans les espèces de TDP-43 déposées (Scotter, 2015; Wu, 2024):
- Les fragments tronqués C-terminaux sont enrichis dans le cerveau
- La TDP-43 pleine longueur prédomine dans les inclusions de la moelle épinière
Une perte partielle de la fonction TDP-43 dans les modèles murins suffit à provoquer une neurodégénérescence, un dysfonctionnement moteur progressif, une paralysie et, finalement, la mort (Xu, 2014; Yang, 2014).
Voir: Modèle murin TDP-43 de la sclérose latérale amyotrophique (SLA)
DFT
La DFT, également connue sous le nom de dégénérescence lobaire fronto-temporale (DLFT) d'un point de vue pathologique, se caractérise par la présence d'agrégats de protéines TDP-43, tau ou FUS dans les neurones et les cellules gliales (Prasad, 2019; Ho, 2024; Hu, 2024).
- Les mutations TDP-43 ne sont associées qu'à un faible pourcentage de cas de DFT.
- Par exemple, la mutation C9orf72 représente environ 13 % des cas de DFT.
- Cependant, des agrégats de TDP-43 sont présents chez jusqu'à 50 % des patients atteints de DFT.
- Une autre mutation importante est celle du gène GRN (progranuline), qui entraîne une perte de fonction et se transmet de manière autosomique dominante.
Les symptômes de la DFT liés à la TDP-43 comprennent des changements de personnalité, de comportement et de compétences linguistiques, tels que la perte d'empathie, un comportement social inapproprié et des difficultés de communication (Prasad, 2019; de Boer, 2020; Jo, 2020).
Consultez notre ressource: Neuroimagerie dans la démence frontotemporale et essais cliniques
Chez les patients atteints de DFT, les taux sériques totaux de TDP-43 sont souvent nettement inférieurs à ceux des témoins sains, en particulier chez les porteurs de l'expansion répétitive C9orf72 ou chez ceux atteints de DFT avec un phénotype concomitant de maladie des motoneurones. Cette diminution des taux serait due au fait que la protéine est séquestrée dans des agrégats insolubles (Katisko, 2022).
Trois types différents de pathologies ont été rapportés dans la DFTF : la DFTF-tau (35-50 %), la DFTF-FUS (10 %) et la DFTF-TDP (environ 50 %). La DFTF-TDP est divisée en plusieurs sous-types (Prasad, 2019; de Boer, 2020; Meneses, 2021) :
- Type A: inclusions cytoplasmiques neuronales (ICN) compactes ou en forme de croissant, généralement présentes dans le néocortex.
- Type B: NCI diffuses ou granulaires, souvent localisées dans les oligodendrocytes corticaux et sous-corticaux, et associées à une maladie des motoneurones.
- Type C: nombreuses neurites dystrophiques, principalement dans le néocortex.
- Type D: nombreuses inclusions intranucléaires neuronales.
Veuillez consulter nos innovations: IRM de diffusion et démence frontotemporale (DFT) et démence frontotemporale (DFT) et IRM de l'atrophie cérébrale.
La TDP-43 constitue-t-elle une cible thérapeutique potentielle?
Les stratégies thérapeutiques actuelles mettent l'accent sur trois approches principales : éliminer la protéine toxique, restaurer sa fonction et traiter les effets en aval de sa déplétion.
Anticorps vectorisés
- L'ACI-5891 est un anticorps monoclonal vectorisé administré à l'aide de l'AAV9, qui cible spécifiquement la TDP-43 pathologique. Dans des modèles murins de SLA et de DFT, une seule injection intracisternale a permis de réduire jusqu'à 68 % les signaux pathologiques de la phospho-TDP-43 (Val, 2025).
- L'anticorps 3B12A détecte l'exposition anormale des résidus E246 et D247 dans les agrégats de TDP-43 mal repliés et est conçu pour favoriser la dégradation de la TDP-43 par le protéasome (Francois-Moutal, 2021).
- Le VH7Vk9 favorise la dégradation par le protéasome ou l'autophagie en se concentrant sur le domaine RRM1 (Francois-Moutal, 2021).
Petites molécules
- La baicaléine agit comme un agent correcteur de structure, aidant à transformer les protéines mal repliées existantes en formes fonctionnelles (Chang, 2024).
- Le M102 est un activateur des voies du facteur nucléaire érythroïde 2 (NRF2) et du facteur de choc thermique 1 (HSF-1), qui s'avère prometteur dans les modèles animaux de la SLA en réduisant le stress oxydatif et les effets de l'agrégation de la TDP-43 (San Gil, 2025).
- D'autres molécules ont été étudiées dans des modèles de mouches (Francois-Moutal, 2021) :
- rTRD0, qui cible l'interface de liaison de l'acide nucléique RRM1
- nTRD22, qui cible le domaine N-terminal pour favoriser l'élimination de la TDP-43
Oligonucléotides antisens (ASO)
- Le QRL-201 cible la STMN2, une protéine mal épissée dans la pathologie TDP-43, ce qui contribue à améliorer la fonction des motoneurones. Ce traitement fait actuellement l'objet d'essais de phase 1 pour la SLA sporadique et la SLA C9orf72 (Liu, 2024).
- Qalsody est approuvé par la FDA pour les patients atteints de SLA présentant une mutation SOD1, mais ne traite pas la pathologie TDP-43 (San Gil, 2025).
Thérapies géniques
- En cours de développement préclinique pour corriger l'épissage aberrant d'autres gènes affectés, tels que UNC13a (San Gil, 2025).
Thérapies ciblant les chaperons
- La surexpression des protéines du domaine J (DNAJB1, DNAJB2a, DNAJB4, DNAJB5) réduit considérablement la TDP-43 insoluble dans les modèles cellulaires (San Gil, 2025).
14-3-3θ
- Cible la TDP-43 mal repliée dans des modèles murins de SLA, ce qui entraîne une amélioration des déficits comportementaux (San Gil, 2025).
Induction de l'autophagie
- Améliore l'élimination des agrégats protéiques (Hayes, 2022) :
- Inhibiteurs mTOR tels que la rapamycine
- Les inducteurs TFEB tels que le tréhalose et l'ibudilast
- Veuillez consulter nos ressources : Altération de l'autophagie microgliale dans les maladies neurodégénératives et autophagie et facteur de transcription EB (TFEB)
Le domaine C-terminal riche en glycine et le domaine RRM2 jouent un rôle crucial dans la séparation de phase et l'agrégation, ce qui en fait des cibles intéressantes pour les anticorps spécifiques (Riemenschneider, 2023). Comme l'ont indiqué les auteurs, la prudence est de mise lorsqu'on cible des domaines essentiels, car l'immunisation active avec certains peptides N-terminaux a entraîné la mort de souris, probablement en raison du rôle physiologique de la TDP-43 dans le noyau (Riemenschneider, 2023).
Plusieurs groupes de recherche ont démontré une modification de la maladie grâce à des interventions thérapeutiques sur des modèles murins de SLA TDP-43 ΔNLS. Parmi les résultats notables, on peut citer :
- AIT-101: une petite molécule qui inhibe PIKfyve. Ce traitement s'est avéré réduire la perte de poids corporel, améliorer les déficits moteurs et diminuer les niveaux de neurofilament léger (NfL) dans le plasma et le LCR, ainsi que réduire les agrégats de TDP-43 (Young, 2023).
- VRG50304: il a été démontré que ce composé réduit les niveaux de NfL dans le plasma et le LCR, ainsi que l'activité de la sphingomyélinase acide dans le cortex causal (Stomakhina, 2021).
- AAV9/NF242: Les injections intracérébroventriculaires du fragment N-terminal du facteur d'échange de nucléotides guanine rho ont amélioré à la fois la durée de vie et les performances motrices des souris (Droppelmann, 2024).
Veuillez consulter notre ressource pour plus de détails : Souris TDP-43 ΔNLS (rNLS8) pour le développement de médicaments contre la SLA
Bien que plusieurs thérapies ciblant la TDP-43 aient échoué dans les essais cliniques sur la SLA, telles que l'arimoclomol, le phénylbutyrate de sodium et l'ursodoxicoltaurine, la colchicine et le BIIB105 (San Gil, 2025), la TDP-43 reste une cible importante dans les recherches en cours. Bien qu'aucun traitement basé sur la TDP-43 n'ait encore démontré avec succès sa capacité à ralentir la progression de la maladie chez l'homme, de nombreux traitements sont en phase de développement préclinique, ce qui constitue une base solide pour les progrès futurs. Pour progresser vers des traitements efficaces, il faudra mettre en place des stratégies innovantes qui s'attaquent à la fois à l'agrégation de la TDP-43 et à sa perte fonctionnelle, ouvrant ainsi la voie à des avancées potentielles dans le traitement de la SLA (San Gil, 2025).
Notre équipe se fera un plaisir de répondre à toutes vos questions concernant le TDP-43 ou de vous fournir des informations spécifiques sur les modèles que nous utilisons pour les études d'efficacité thérapeutique.
En savoir plus sur nos modèles de maladies neurodégénératives
Contenu connexe
Informations actualisées sur la sclérose en plaques et les meilleures pratiques relatives à l'évaluation des agents thérapeutiques dans les modèles animaux de la SEP.
Interactions entre les microglies et les neurones et maladies neurodégénératives
Une revue concise des interactions directes entre les microglies et les neurones, et de la manière dont ces interactions intercellulaires peuvent être affectées dans les maladies neurodégénératives.
Modèles de souris SLA pour le développement de médicaments
Un guide pour l'utilisation la plus efficace possible des modèles animaux de recherche sur la sclérose latérale amyotrophique (SLA) pour les essais précliniques de produits thérapeutiques.
Souris TDP-43 ΔNLS (rNLS8) pour le développement de médicaments contre la SLA
Cette ressource fournit des informations sur l'utilisation du modèle de souris transgénique TDP-43 ΔNLS (deltaNLS, hTDP-43ΔNLS, hTDP-43DeltaNLS, dNLS, TDP43 NLS, rNLS8) de la SLA pour des études thérapeutiques précliniques.
La neuroimagerie dans la démence frontotemporale et les essais cliniques
L'utilité des biomarqueurs d'imagerie IRM et TEP dans notre compréhension des variantes de la démence frontotemporale (DFT) et leur utilisation comme critères d'évaluation dans les essais cliniques sur la DFT.
IRM de diffusion et démence frontotemporale (DFT)
Analyse de la neuroimagerie de diffusion à partir de l'étude FTLDNI sur l'histoire naturelle de la démence frontotemporale (DFT)
Démence frontotemporale (DFT) et atrophie cérébrale par IRM
Biomarqueurs IRM (y compris l'atrophie cérébrale) issus des études FTLDNI sur l'histoire naturelle de la démence frontotemporale (DFT).