Impaired Microglia Autophagy in Neurodegenerative Diseases
How impaired microglial autophagy contributes to the progression of neurodegenerative diseases.
What is microglia autophagy?
Microglia are the resident immune cells of the central nervous system (CNS) and are essential for maintaining brain homeostasis. These highly dynamic cells help regulate the blood-brain barrier (BBB), prune synapses, and coordinate immune responses. By continuously surveying their environment, microglia detect and eliminate pathogens, cellular debris, and protein aggregates, often through receptors, such as toll-like receptors (TLRs).
When neurotoxic protein aggregates accumulate, they stimulate the release pro-inflammatory cytokines like interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α). This situation creates a pro-inflammatory environment that accelerates further protein misfolding and aggregation, fueling a vicious cycle of neuroinflammation and neuronal damage. Although most research has focused on neurons, protein aggregates also accumulate in glial cells, including microglia, implicating them in neurodegenerative pathology.
Autophagy and phagocytosis are two key cellular degradation processes. Phagocytosis mainly clears extracellular debris, while autophagy targets intracellular components, such as damaged organelles and misfolded proteins, directing them to lysosomes for degradation. In microglia, autophagy contributes not only to intracellular quality control, but also to the regulation of immune activity. This function becomes increasingly important with aging, as autophagy efficiency declines – a recognized hallmark of aging.
Autophagy has three primary forms (Jülg, 2021):
- Microautophagy involves the direct engulfment of cytosolic material by lysosomes
- Chaperone-mediated autophagy selectively targets specific proteins based on chaperone recognition
- Macroautophagy, the most widely studied type, involves the formation of double-membraned autophagosomes that engulf cellular components and subsequently fuse with lysosomes to form autolysosomes for degradation.
Macroautophagy is tightly regulated by autophagy-related (ATG) proteins. This process includes activation of the ULK1 complex, formation of the Beclin1-PIK3C3 complex, and conversion of LC3-I to LC3-II (Jülg, 2021). Key regulators include the mammalian target of rapamycin complex 1 (mTORC1), which inhibits autophagy, and AMP-activated protein kinase (AMPK), which promotes it. Proper autophagy depends on maintaining a balance between these opposing signals (Lin, 2023; Ou-Yang, 2023). In microglia, additional regulatory pathways include p38 MAPK signaling and adaptor protein p62, often used as a marker of autophagic impairment (Lin, 2023; Ou-Yang, 2023).
Emerging evidence highlights the role of microglial autophagy in regulating CNS inflammation (Zhu, 2022). When autophagy is disrupted, essential microglial functions, such as phagocytosis and immune regulation, are impaired, resulting in chronic neuroinflammation (Zhu, 2022). Since neuroinflammation is a defining feature of many neurodegenerative diseases, autophagy acts as a protective mechanism by clearing inflammatory mediators before they reach harmful levels. When this system fails, persistent inflammation and neuronal injury follow (Lin, 2023).
A central anti-inflammatory function of microglial autophagy is the suppression of the NLRP3 inflammasome, a multiprotein complex that activates pro-inflammatory cytokines. Impaired autophagy leads to increased NLRP3 activation, contributing to neurotoxicity. This relationship is reciprocal – NLRP3 activity can also disrupt autophagy, forming a feedback loop that plays a key role in CNS immune dysregulation.
See: What is NLRP3? & What is an inflammasome?
Although neuron-focused studies have dominated autophagy research, increasing data support the view that microglial autophagy dysfunction contributes significantly to neurodegenerative diseases. Enhancing autophagic activity in microglia is now under investigation as a therapeutic approach. By improving the clearance of misfolded proteins and mitigating chronic inflammation, restoring autophagy in microglia may slow or prevent the progression of disorders driven by both protein aggregation and immune imbalance.
How is microglia autophagy impaired in neurodegenerative disease, such as AD, PD, and ALS?
Alzheimer's Disease (AD)
AD, the most prevalent neurodegenerative disease, is characterized by progressive cognitive decline and the accumulation of extracellular amyloid-beta (Aβ) plaques and intracellular tau tangles. Microglia have a dual role in AD, participating in both the clearance of pathological aggregates and also contributing to inflammation (Cho, 2014).
Key autophagy-related regulators implicated in microglial function in AD include:
- PPAR-α
- Atg1
- Map1lc3b
- BECN1
Activation of PPAR-α enhances autophagy in human microglia and AD mouse models, leading to reduced Aβ plaque burden, better cognitive performance, and increased glial activity near plaques (Luo, 2020). Conversely, knocking down Atg1 or Map1lc3b impairs Aβ degradation and heightens inflammatory signaling in experimental models (Cho, 2014)
Partial loss of BECN1 function in mice leads to elevated microglial NLRP3 inflammasome activity and increased production of IL-1β and IL-18 (Houtman, 2019). In APP/PS1 mice with reduced BECN1 expression, greater NLRP3 and caspase-1 activation is observed. Super-resolution imaging studies confirm colocalization of NLRP3 with LC3-positive vesicles, suggesting that autophagy may directly degrade inflammasome components (Houtman, 2019).
These findings are supported by human data. Microglia isolated from AD patients show reduced Beclin1 levels, which likely contributes to impaired Aβ clearance due to compromised autophagosome formation and phagocytic function (Lucin, 2013). After Aβ phagocytosis, microglia degrade it through a mechanism involving LC3-II and the adaptor protein optineurin (OPTN), regulated by the STK11/LKB1–AMPKα pathway. These insights reinforce the essential role of intact autophagy in limiting Aβ accumulation and inflammation in AD.
Parkinson's Disease (PD)
PD is the second most common neurodegenerative disease, marked by the progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc). Clinically, PD manifests with motor symptoms, such as tremor, rigidity, bradykinesia, and postural instability, along with non-motor features including cognitive decline and sleep disturbances. The pathological hallmark is the accumulation of α-synuclein in Lewy bodies and neurites.
Microglial autophagy is key to regulating inflammation and clearing α-synuclein. Excessive α-synuclein disrupts autophagic flux and induces oxidative stress. The DJ-1 gene, linked to familial PD, regulates microglial autophagy. Its deletion impairs α-synuclein clearance and amplifies inflammation (Nash, 2017). Deleting Atg5 in microglia increases NLRP3 activation and worsens neuronal loss in the MPTP model (Qin, 2021). Inhibiting Drp1 pharmacologically restores autophagy and reduces α-synuclein accumulation (Fan, 2019).
Extracellular α-synuclein inhibits autophagy through TLR4-mediated p38 and Akt/mTOR signaling pathways. Blocking any of these pathways helps restore autophagic function (Tu, 2021). Microglia also engage in “synucleinphagy,” a selective autophagic process for degrading neuron-released α-synuclein, dependent on TLR4 signaling and NF-κB-driven p62 induction (Choi, 2020).
Aging worsens these impairments. Aged mice retain α-synuclein longer than young mice following injection of human α-synuclein, pointing to an age-related decline in autophagic efficiency that contributes to disease progression (Hong, 2024).
Amyotrophic Lateral Sclerosis (ALS)
ALS is a progressive and fatal neurodegenerative disease characterized by motor neuron loss. It features cytoplasmic inclusions of TDP-43 and is genetically linked to mutations in C9ORF72 and SOD1. Microglial activation and dysfunction are prominent in ALS pathology, although autophagy’s specific role is less well understood.
Microglia-like cells derived from human induced pluripotent stem cells (hiPSC-MG) with PFN1 mutations exhibit impaired autophagy and phagocytosis (Funes, 2024). These deficits are reversed by rapamycin, indicating an autophagy-dependent mechanism (Funes, 2024). hiPSC-derived microglia with C9ORF72 mutations show reduced autophagy and increased immune activity, while pharmacologically stimulating autophagy improves motor neuron survival (Banerjee, 2023).
In SOD1G93A ALS mice crossed with LC3 reporter lines, microglial and astrocytic flux increases during symptomatic phases, though this response is absent in oligodendrocytes (Perera, 2025). Moreover, autophagic dysfunction appears earlier in the spinal cord than in the motor cortex, indicating regional and cell-type specificity in disease progression (Perera, 2025).
Together, evidence from AD, PD, and ALS suggests a shared mechanism in which impaired microglial autophagy promotes protein accumulation and chronic inflammation, accelerating neuronal degeneration. Enhancing autophagy in microglia may therefore represent a viable strategy to slow disease progression.
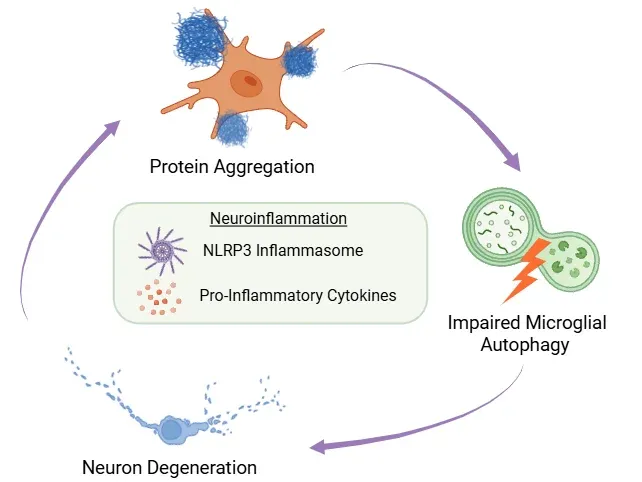
Neuroinflammatory Feedback Loop.
Misfolded protein buildup, impaired microglial autophagy, and neuronal degeneration form a self-reinforcing cycle. Neuroinflammation - driven by NLRP3 inflammasome activation and cytokine release - is central to this process.
How can microglia autophagy be targeted therapeutically for neurodegenerative diseases?
Targeting microglial autophagy offers a promising therapeutic avenue for treating neurodegenerative diseases such as AD, PD, and ALS. Although no currently approved treatments selectively modulate microglial autophagy, increasing preclinical evidence supports the idea that enhancing autophagic activity can improve clearance of pathological proteins, reduce inflammation, and promote CNS homeostasis.
Several pharmacological agents have been investigated:
- Rapamycin, an mTOR inhibitor induces autophagy and reduces TLR2-mediated α-synuclein expression (Dzamko, 2017). However, in ALS models, rapamycin exacerbated motor neuron loss (Zhang, 2011), and clinical trials have yet to demonstrate significant benefits (Mandrioli, 2023).
- Metformin, which activates AMPK, has shown neuroprotective effects in PD models by reducing α-synuclein aggregation, though its direct impact on microglial autophagy remains unclear (Lu, 2016).
- Fluoxetine enhances microglial autophagy by increasing LC3-II levels and autophagosome formation, with potential for reducing inflammation and clearing aggregates (Park, 2021).
- Kaempferol boosts autophagy in microglia and suppresses NLRP3 activation, reducing cytokine secretion in microglia (Han, 2019).
- Trehalose, an autophagy inducer, was promising in early ALS studies, but recent clinical trial results were inconclusive (HEALEY ALS Platform Trial, 2025).
Given these mixed outcomes, researchers are developing cell type-specific delivery strategies:
- Nanoparticles that cross the BBB and target microglia, such as MCPZFS NPs, have shown the ability to enhance Aβ clearance and reduce cytokine release. These particles may redirect protein degradation from autophagy to the proteasomal pathways (Liu, 2019). However, challenges such as BBB penetration, targeting precision, and nanoparticle variability remain (Lin, 2023).
- Graphene oxide (GO), a carbon-based nanomaterial, promotes autophagy by inhibiting mTOR signaling through AMPK activation. GO has been shown to enhance microglial clearance of Aβ and reduce neurotoxicity in disease models (Li, 2020).
Gene therapy approaches are also under exploration:
- Adeno-associated virus (AAV) vectors with glial cell-specific promotors allow targeted regulation of autophagy genes in microglia (O’Carroll, 2021). This approach offers advantages over systemic drugs, especially considering the difficulty of crossing the BBB.
Combining autophagy inducers with inflammasome inhibitors may yield synergistic benefits. Co-targeting both pathways has shown more effective suppression of protein accumulation and inflammation compared to either intervention alone (Wang, 2023).
In summary, while most strategies for modulating microglial autophagy are still in early development, the field is rapidly progressing. Continued work is needed to refine delivery systems, enhance cell-specificity, and establish long-term safety. Therapies that restore autophagy in microglia hold significant promise for slowing neurodegeneration by addressing both proteinopathy and immune dysregulation.
Our team would be happy to answer any questions about impaired microglial autophagy in neurodegenerative diseases or provide specific information about the models we use for therapeutic efficacy studies.
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on Neuroinflammation and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
Lysosome Dysfunction in Microglia & Astrocytes
An overview of lysosomal dysfunction in microglia & astrocytes, and its role in neurodegenerative diseases.
Autophagy, Parkinson's Disease, and Dopaminergic Neurons
An overview of how impaired autophagy can lead to pathologic changes and neurodegeneration in dopaminergic neurons in Parkinson’s disease.
Autophagy & Neurodegenerative Diseases
An overview of how cellular autophagy plays a role in brain health and neurodegeneration.
NLRP3 Inflammasome and Neurodegenerative Diseases
An overview of the NLRP3 inflammasome and its role in neurodegenerative diseases, including Alzheimer's disease, Parkinson’s disease, and ALS.
TNF-α (TNF-alpha) & Microglia in Neurodegenerative Diseases
An overview of the function of tumor necrosis factor-alpha (TNF-α) in microglia and its contribution to the progression of neurodegeneration.
Autophagy and Transcription Factor EB (TFEB)
An overview of Transcription Factor EB (TFEB) and its role in autophagy and neurodegenerative diseases.