Microglia, Astrocytes & Tau in Neurodegenerative Diseases
How glial-driven neuroinflammation fuels tau aggregation, propagation, and neuronal loss in Alzheimer’s disease and other tauopathies.
This resource describes:
- How do microglia and astrocytes interact in neurodegenerative diseases?
- How does neuroinflammation intersect with tauopathies?
- How do microglia contribute to tau pathology in neurodegenerative diseases?
- How do astrocytes amplify and convey tau pathology?
- Can glial-mediated clearance of tau aggregates be enhanced therapeutically?
How do microglia and astrocytes interact in neurodegenerative diseases?
Microglia and astrocytes are two key types of glial cells that play essential roles in maintaining homeostasis and responding to injury within the central nervous system (CNS). Both cell types contribute to the brain's immune response and are critical players in neuroinflammation, a common feature in neurodegenerative diseases.
Microglia
Microglia are the primary immune cells in the CNS, derived from yolk sac macrophages and distinct from peripheral immune cells (Ginhoux, 2010; Leavy, 2010). They are highly dynamic and constantly monitor the microenvironment of the brain using their specialized receptor repertoire (Qin, 2023). Microglia are crucial for phagocytosis, synaptic pruning, and secretion of signaling molecules that regulate neuronal functions (Gao, 2023).
Recent advances have revealed a wide range of microglial states, moving beyond the traditional binary classification of M1 (pro-inflammatory) and M2 (anti-inflammatory) microglia. Instead, microglia exhibit a spectrum of functional phenotypes that vary depending on environmental cues, genetic factors, and disease progression. For instance, in Alzheimer’s Disease (AD), disease-associated microglia (DAM) are localized near amyloid plaques and contribute to the clearance of amyloid-β aggregates (Keren-Shaul, 2017; Gao, 2023). Conversely, sustained activation of microglia can lead to excessive cytokine production, impairing synaptic function and exacerbating neuronal damage (Kwon, 2020). Microglia also release extracellular vesicles that contribute to neurodegeneration by spreading misfolded proteins, such as tau and amyloid-beta in AD and alpha-synuclein in Parkinson’s Disease (PD) (Gao, 2023).
Astrocytes
Astrocytes are the most abundant neural cell in the CNS, and are critical for maintaining homeostasis, regulating blood flow, and supporting synaptic transmission. They are actively involved in modulating the blood-brain barrier (BBB) and are a key component of the glymphatic system, play a major role in synapse and neuronal function, and controlling the extracellular balance of ions and neurotransmitters and removing dead cells (Abbott, 2010; Sweeney, 2018; Kwon, 2020).
Astrocytes, like microglia, can exist in reactive states that range from protective to harmful. Pro-inflammatory astrocytes (A1 phenotype) release cytokines, such as IL-1β, TNF-α, and complement cascade proteins, which are known to damage neurons and synapses. Conversely, anti-inflammatory astrocytes (A2 phenotype) promote neuronal survival and repair by secreting neurotrophic factors and other protective molecules. Astrocytic dysfunction is commonly observed in neurodegenerative diseases, often exacerbating neuroinflammation and contributing to disease progression (Kwon, 2020).
In AD, for instance, astrocytic activation is associated with impaired glutamate clearance, leading to excitotoxicity and neuronal death. Moreover, reactive astrocytes induced by microglial cytokines can compromise the BBB, further aggravating neuronal vulnerability (Kwon, 2020). Similarly, in ALS, reactive astrocytes expressing toxic factors, such as C3, contribute to motor neuron degeneration (Gao, 2023).
Microglia-Astrocyte Crosstalk
Microglia and astrocytes are vital to the maintenance of CNS homeostasis and play crucial roles in responding to injury and disease. Their interactions are highly dynamic and can influence neuroinflammation and neurodegeneration in either protective or harmful ways, depending on their state and environmental context (Liu, 2020; Kim, 2021; Gotoh, 2023). In response to pathological stimuli, microglia become activated and release pro-inflammatory factors, such as IL-1α, TNF-α, and C1q. These molecules drive astrocytes toward a reactive A1 phenotype, characterized by the release of additional pro-inflammatory cytokines that sustain microglial activation. This reciprocal signaling establishes a damaging feedback loop that amplifies inflammation and accelerates neurodegenerative processes (Gao, 2023). In this reactive state, microglia also increase synaptic pruning, a process normally essential for neural circuit refinement, but one that can become harmful under inflammatory conditions. Concurrently, astrocytes stimulate neurons to release complement proteins, which further activate microglia to phagocytose synapses and even entire neurons, contributing to neuronal loss (Sierra, 2013). This escalating cycle of inflammation can disrupt the blood-brain barrier (BBB), weakening its protective function and allowing peripheral immune cells, such as B cells, T cells, and monocytes, to infiltrate the CNS. These infiltrating cells exacerbate inflammation by enhancing cytokine release from glial cells, creating an environment of sustained immune activation that can further damage neural tissue (Abbott, 2010; Sweeney, 2018).
Despite these harmful interactions, microglia and astrocytes are also capable of promoting protective and regenerative processes. Under certain conditions, microglia can induce astrocytes to adopt the anti-inflammatory A2 phenotype through IL-10 signaling, fostering a more restorative environment that limits tissue damage and promotes repair (Mohammad, 2024). Following CNS injury, astrocytes increase their proliferation and form a "glial scar," a physical barrier that helps contain the injury site and prevents further tissue damage. While this scar can sometimes inhibit neural regeneration, it plays a critical role in stabilizing the CNS environment during the early stages of recovery. Beyond scar formation, both microglia and astrocytes release factors that support extracellular matrix (ECM) reconstruction, neovascularization, and angiogenesis. These processes create a favorable environment for neuronal survival, encourage the migration of stem cells to replace damaged neurons, and facilitate axonal remodeling to restore lost connections (Yang, 2019; Heithoff, 2021).
Ultimately, the interactions between microglia and astrocytes are complex and tightly regulated, with outcomes that can vary widely depending on the balance between pro-inflammatory and anti-inflammatory signals. While their interplay has the potential to drive damaging inflammation and neuronal loss, it is equally critical for promoting repair and restoring CNS homeostasis following injury or disease. Understanding these dual roles is essential for developing strategies to mitigate neurodegenerative damage while enhancing the CNS's intrinsic regenerative capacity.
How does neuroinflammation intersect with tauopathies?
Tauopathies are a group of neurodegenerative disorders characterized by the progressive accumulation of hyperphosphorylated tau protein into intracellular fibrillar aggregates. These aggregates, which vary in isoform composition, structural conformation, and localization, are most commonly deposited within neurons. Tauopathies are classified into primary forms, where tau deposition is the predominant pathological hallmark, e.g. progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and frontotemporal dementia (FTD); secondary forms, which feature additional pathological contributions, such as amyloid-beta in Alzheimer’s disease (AD); and geographic forms, like Guadeloupean parkinsonism (Lannuzel, 2007). During disease progression, tau proteins undergo post-translational modifications that cause them to misfold into β‑sheet‑rich aggregates, which then propagate from their original deposition site to anatomically connected regions through a prion-like mechanism. This process involves tau proteopathic seeds recruiting tau monomers (Jucker, 2018) . While glial cells, including microglia and astrocytes, initially attempt to mitigate the damage by phagocytosing extracellular tau and releasing protective factors, chronic exposure leads them into reactive states. These reactive states are characterized by the activation of NF‑κB pathways, inflammasome-driven transcription, excessive cytokine release, impaired lysosomal clearance, and errors in synaptic pruning. Such sustained neuroinflammatory conditions promote tau seeding, hasten neuronal dysfunction, and tightly correlate with cognitive decline, further aggravating the progression of tauopathies.
How do microglia contribute to tau pathology in neurodegenerative diseases?
There is increasing agreement that both pro-inflammatory and hypofunctioning microglia play distinct roles in contributing to tau pathology and neurodegeneration (Streit, 2014; Angelova, 2019; Odfalk, 2022). Odfalk very succinctly identified this paradigm in the recent review entitled Microglia: Friend and foe in tauopathy (Odfalk, 2022). During the earliest stages of disease, microglia fulfil a protective role. They migrate toward dystrophic neurites, engulf soluble and filamentous tau released from dying neurons, and secrete anti‑inflammatory mediators that help re‑establish tissue homeostasis. Yet, this beneficial surveillance state is fragile. Persistent proteopathic stress, age‑associated priming, genetic risk alleles, and/or comorbid cerebrovascular insults drive microglia toward a disease‑associated microglia (DAM) phenotype. DAMs down‑regulate homeostatic markers, while up‑regulating phagolysosomal and lipid‑metabolism genes that paradoxically enhance the uptake, intracellular fragmentation, and subsequent release of more seeding‑competent tau species.
There is substantial evidence of the positive role of microglia phagocytosis in the clearance of tau (Luo, 2015; Bolós, 2017; Leyns, 2017). Tau pathology has been experimentally shows to improve when neuroinflammation is reduced by altering microglia phenotype, either through TNFα suppression (Gabbita, 2015) or IL-1 signaling blockade in 3xTgAD mice (Kitazawa, 2011). There is also evidence microglia contribute directly to the progression of tau pathology through various mechanisms including dysregulation of their homeostatic functions (Perea, 2018; 2020). For instance, microglia can facilitate the intercellular transmission of tau seeds that subsequently form aggregates. This mechanism is supported by Asai et al., who demonstrated that the depletion of microglia reduced tau propagation (Asai, 2015). Additionally, there is compelling evidence that plaque-associated microglia promote tau propagation in transgenic mouse models (Clayton, 2021).
An illustrative example of the normal versus pathological balance that occurs within the context of tauopathies, is the role of the fractalkine receptor CX3CR1 (Perea, 2018). Under normal conditions, microglia remain in a resting state, where the CX3C Ligand (CX3CL1) expressed on neurons (Jiang, 2023) and the receptor (CX3CR1) expressed on microglia (Bhaskar, 2010; Zhan, 2014; Maphis, 2015) interact to maintain the normal functionality. This axis is maintained through the binding of membrane-bound and soluble forms of CX3CL1 to the microglial receptor CX3CR1, ensuring proper immune function. When extracellular tau is present, microglia become activated and attempt to clear the excess tau through phagocytosis, a process influenced by the CX3CL1/CX3CR1 axis. In Alzheimer’s disease, as pathology progresses and neuronal death rises, there is a notable accumulation of extracellular tau, which interferes with the CX3CL1/CX3CR1 pathway's normal function. The excessive tau competes with CX3CL1 for binding to CX3CR1, thereby impairing microglial clearance of tau (Bolós, 2017). In response, microglia become hyperactivated, proliferate, and attempt to compensate by producing more CX3CL1 and CX3CR1. Despite this increase, the CX3CL1/CX3CR1 axis becomes dysregulated, further contributing to tau accumulation and disease progression. This dysfunctional microglial response highlights the complex role of immune regulation.
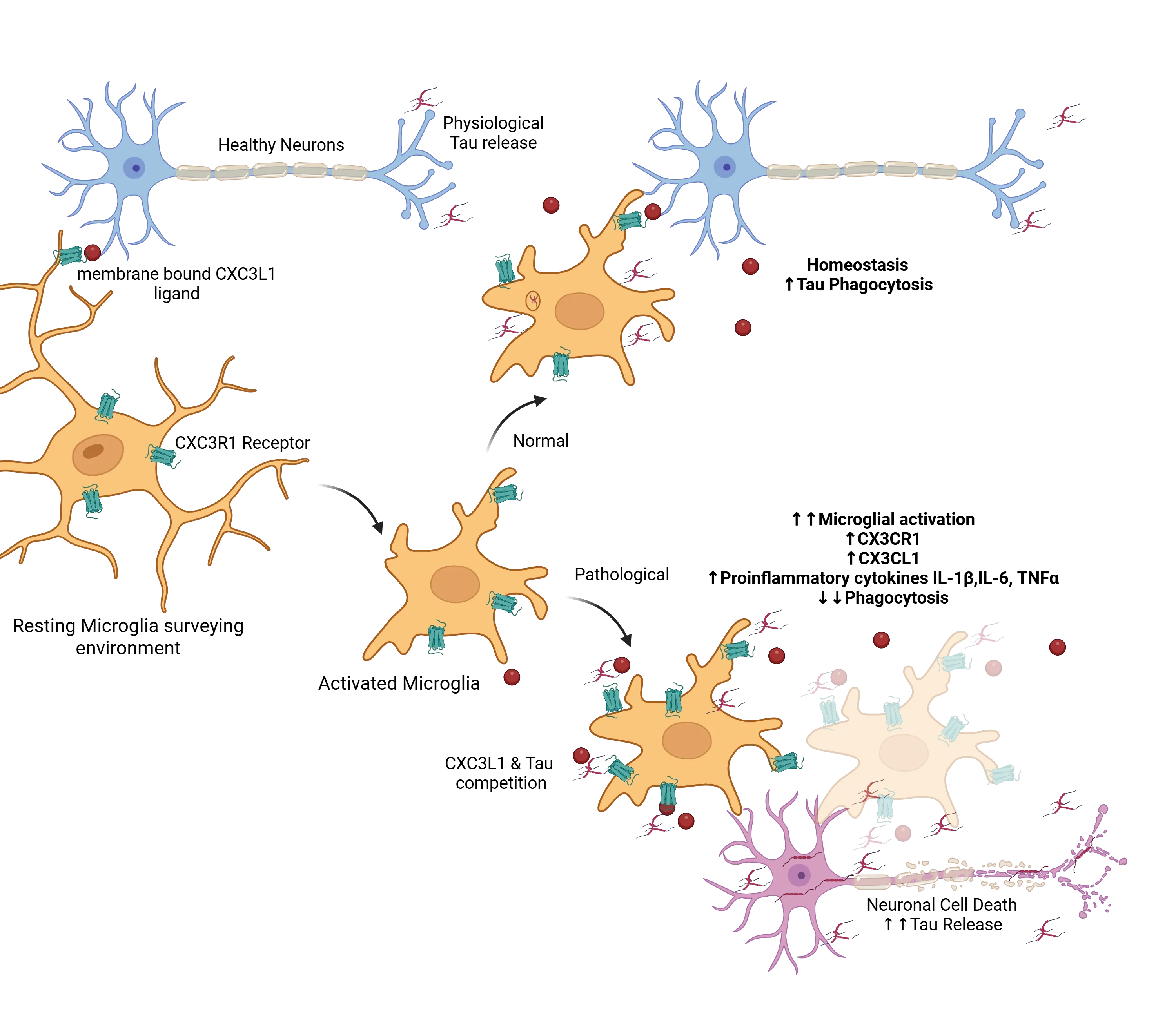
Signals such as the CXCL1/CXC3R1 help maintain a resting state in microglia as they survey their environment. In the presence of extracellular tau, these microglia are activated and under normal conditions will phagocytose the tau and help maintain homeostasis. Under pathological conditions, excess tau competes with the CXCL1/CXC3R1 interactions, pushing the activated microglia into dysfunctional state, with increased activation, decreased phagocytosis, reduced tau clearance and consequently increase extracellular tau accumulation, which exacerbates the toxic cycle. Figure reproduced from Perea et al. (Perea, 2018) under the Creative Commons Attribution License.
How do astrocytes amplify and convey tau pathology?
Astrocytes have increasingly been recognized as central players in tau pathology, not merely as passive components affected by neuronal dysfunction, but as active contributors to both the regulation of tau homeostasis and its pathological spread across brain regions. Emerging research indicates that astrocytes engage in complex mechanisms to internalize tau, influencing its accumulation and clearance. These glial cells exhibit a dual role: on one hand, they can act as buffers by attempting to mitigate the extracellular tau burden, while on the other, their dysfunction or overactivation may amplify tau aggregation and propagation. This nuanced role highlights astrocytes as potential targets for therapeutic interventions aimed at modulating tau dynamics in neurodegenerative diseases (Reid, 2020).
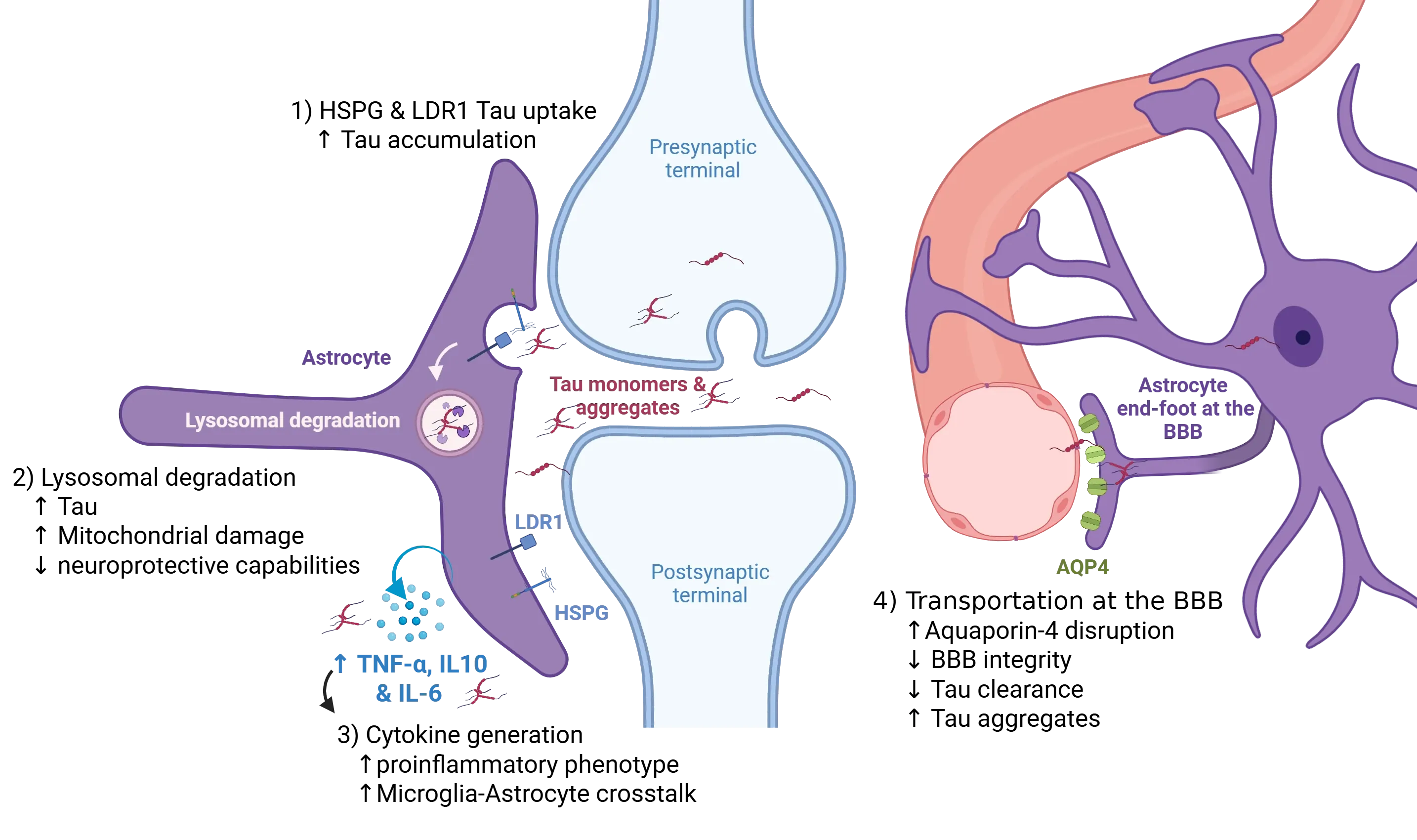
Astrocytes play an active role in modulating the propagation of tau pathology within the central nervous system. Neurons release tau species into the extracellular space through multiple pathways, including vesicular secretion and synaptic release at the pre-synapse. Perisynaptic astrocytes internalize extracellular tau via receptor-mediated endocytosis, primarily through specific heparan-sulfate proteoglycans (HSPGs), which preferentially bind aggregated tau, and the low-density lipoprotein receptor-related protein 1 (LRP1), which mediates uptake of both monomeric and oligomeric tau (1). Disruption of these pathways, can alter the balance between clearance and accumulation of extracellular tau, potentially enhancing tau spread. Once internalized, tau is trafficked to lysosomes for degradation (2); however, impairment of lysosomal function, due to intrinsic overload or pathological signalling, leads to intracellular tau accumulation and the formation of seeding-competent aggregates. In parallel, a pro-inflammatory milieu, marked by elevated cytokines, such as tumor necrosis factor-alpha (TNF-α) and the interleukins IL-10 & IL-6, amplifies astrocytic reactivity and facilitate astrocyte-microglia crosstalk (3), thereby increasing the pool of pathogenic tau species available for propagation. Furthermore, disruption of aquaporin-4 (AQP4) localization at perivascular astrocytic end-feet compromises glymphatic clearance of tau (4). Disruption of this perivascular clearance mechanism results in the progressive accumulation of extracellular tau, thereby exacerbating the spread of tau pathology throughout the brain parenchyma. Figure derived from Reid et al. (Reid, 2020) under the Creative Commons Attribution License.
At the synapse, neurons release both monomeric and aggregated tau around the synaptic cleft, where astrocytes rapidly sequester these species through receptor-mediated endocytosis (Reid, 2020). Astrocytes are uniquely equipped with highly sulfated heparan-sulfate proteoglycans (HSPGs), which exhibit nanomolar affinity for fibrillar tau, directing it efficiently into the endo-lysosomal degradation pathway (Puangmalai, 2020). This capacity is enhanced by the upregulation of the lysosomal regulator TFEB, which expands the astrocytic lysosomal compartment, increasing their ability to process engulfed tau (Martini-Stoica, 2018). Notably, astrocytic tau uptake is not restricted to fibrils. The low-density lipoprotein-related protein-1 (LRP1) works in coordination with HSPGs to internalize both monomeric and oligomeric tau, thus broadening the range of tau species that astrocytes can manage (Reid, 2020). While this astrocytic mechanism initially functions to clear synaptic tau and maintain homeostasis, it paradoxically creates an intracellular reservoir of tau aggregates. These seeding-competent aggregates can be subsequently released, amplifying prion-like propagation and positioning astrocytes as dual actors, both protectors of synaptic tau balance and contributors to its pathologic spread.
Astrocytes at the blood-brain barrier can also play a significant role in tau clearance, particularly through their connection to the glymphatic system. Their perivascular end-feet are enriched with the water channel aquaporin-4 (AQP4), which facilitates the movement of interstitial fluid and drives glymphatic exchange (Sun, 2025). This system helps to clear soluble tau from the brain, thereby mitigating tau pathology. However, disruptions in AQP4 functionality, through multiple mechanisms, including genetic deletion, mislocalization, sleep fragmentation, or aging, can impair glymphatic flow and decrease the BBB integrity, thereby reducing tau clearance and exacerbating extracellular tau accumulation and aggregation. Conversely, interventions aimed at restoring AQP4 polarity or enhancing glymphatic function have shown promise in preclinical studies for reducing tau burden and improving cognitive outcomes (Zhou, 2025). For example, targeted approaches, such as behavioral modifications, pharmacological treatments, and neuromodulatory methods that optimize glymphatic pumping and AQP4 expression provide compelling evidence for therapeutic potential (Sun, 2025).
Can glial-mediated clearance of tau aggregates be enhanced therapeutically?
Enhancing Microglial Clearance of Tau Aggregates
Microglia play a crucial role in clearing misfolded tau proteins. Therapies aimed at enhancing this clearance function are under investigation. One promising avenue involves TREM2 agonism, exemplified by the human monoclonal antibody VG‑3927, which is currently completing Phase 1 dose‑escalation studies. By ligating TREM2, this therapy aims to restore a homeostatic microglial phenotype and boost the phagocytic clearance of extracellular tau. Another approach is NLRP3 inflammasome inhibition, which has shown promise in numerous animal models (Zhang, 2020). Quan showed NLPR3 deficit reduces neuroinflammation and improves cognition in tau transgenic mice (Quan, 2024)
Among the most advanced approaches is TREM2 agonism: the fully human IgG1 antibody VG-3927 is completing first-in-human dose-escalation and pharmacodynamic studies, aiming to boost lipid-sensing pathways, restore microglial metabolism and enhance tau phagocytosis. Earlier antibodies (e.g. Alector’s AL002, Denali’s DNL-919) underperformed in Phase 2 programs, yet an updated TREM2 agonist, Novartis VHB937, entered trials for AD and amyotrophic lateral sclerosis in 2024, underscoring continued optimism in this approach. Complementary to receptor agonism, NLRP3 inflammasome inhibition with the brain-penetrant small-molecule MCC950 suppresses IL-1β maturation, dampening downstream chemokine cascades. Multiple next-generation NLRP3 inhibitors with improved pharmacokinetics have now advanced to IND-enabling studies.
For an in-depth review of the role of TREM2 on microglial function, see: TREM2 and Microglia
Immunotherapy Approaches
Both active and passive immunotherapies targeting tau are under development and hold promise for modulating microglial function by reshaping the extracellular tau landscape (Jadhav, 2019). Active immunotherapies, such as peptide or mRNA vaccines, and passive immunotherapies, including humanized antibodies, aim to selectively deplete soluble tau seeds. By reducing seed-competent conformers or truncated fragments, these therapies can indirectly influence microglial activation, enhancing the uptake and clearance of tau aggregates without provoking excessive neuroinflammation. Second-generation antibodies, designed to target specific pathogenic tau species, are progressing through Phase 1–2 trials, with their success partially dependent on how effectively microglia handle antibody-opsonized tau. These strategies are still in early stages, but ongoing research continues to refine their potential for treating tau-associated neurodegenerative diseases.
Astrocytic Targets: From Glymphatic Flow to Proteostasis
Astrocytes facilitate bulk clearance of interstitial solutes via AQP4-dependent perivascular exchange and secrete heat-shock proteins that chaperone misfolded tau (Zhou, 2025). Experimental up-regulation of AQP4 polarity or pharmacological activation of astrocytic TFEB (a master regulator of lysosomal biogenesis) accelerates tau clearance in murine tauopathy models and is now being explored through small-molecule TFEB agonists and AQP4 modulators (Verkman, 2013). Furthermore, astrocytic conversion of microglial-released cytokines (e.g. IL-33-driven lipid clearance programs) highlights bidirectional glia-glia crosstalk as a potential therapeutic approach (Carlock, 2017; Sun, 2021).
Metabolic Modulation of Tau Post-Translational Processing
Beyond inflammation, metabolic modulation of tau itself remains attractive as a target for therapeutic intervention. Selective inhibition of O-GlcNAcase (OGA) increases O-GlcNAcylation of tau, thereby sterically hindering pathological phosphorylation and aggregation (Kielbasa, 2024; Selnick, 2019; Wang, 2020). MK-8719, an orally available potent and selective small-molecule OGA inhibitor, progressed to Phase 1b studies in PSP; although early efficacy signals were modest, cerebrospinal fluid (CSF) pharmacodynamic biomarkers confirmed robust target engagement. A second OGA inhibitor, ceperognastat (formerly MK-8722), was halted by Eli Lilly in February 2025 after sub-therapeutic exposure in a Phase 2 AD cohort, yet structure and activity refinements are ongoing. Importantly, these metabolic interventions rely on functional glial proteostasis machinery, reinforcing the concept that microglial and astrocytic support is prerequisite for maximal therapeutic benefit.
Outlook
Collectively, data from receptor agonism, inflammasome blockade, immunotherapy, and metabolic modulation converge on a central theme - glial cells possess under-exploited capacity to neutralize pathogenic tau, but this capacity must be re-aligned toward a balanced, homeostatic phenotype. Future trials will need to combine precise molecular engagement with sensitive fluid and imaging biomarkers to capture real-time changes in glial states and tau burden. Whether single-agent glial modulators can yield clinically meaningful slowing of disease progression, or whether they will serve best in rational combination with tau-directed antibodies or antisense oligonucleotides, remains an open question. It is important to note that the field is rapidly evolving, and other therapeutic candidates targeting both astrocytic and microglia-tau interactions are under investigation. Ongoing research continues to shed light on the complex role of glia in tauopathies, aiming to develop effective disease-modifying treatments.
Our team would be happy to answer any questions about microglia, astrocytes & tau in neurodegenerative diseases or provide specific information about the AD and tauopathies models we use for therapeutic efficacy studies.
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on Neuroinflammation and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
TREM2, Microglia and Neuroinflammation
An overview of TREM2, its role in microglia, links to neurodegenerative diseases, and potential treatment implications.
Microglia-Neuron Interactions & Neurodegenerative Diseases
A concise review of the direct interactions between microglia & neurons, and how these cell-to-cell interactions may be affected in neurodegenerative diseases.
Microglia, Astrocytes & α-Synuclein in Parkinson’s Disease
How α-synuclein influences microglia and astrocytes in Parkinson’s disease and other synucleinopathies.
TNF-α (TNF-alpha) & Microglia in Neurodegenerative Diseases
An overview of the function of tumor necrosis factor-alpha (TNF-α) in microglia and its contribution to the progression of neurodegeneration.
Astrocytes & Amyloid-β Mouse Models of Alzheimer's Disease
Analysis of astrocyte morphology in the amyloid-β plaque microenvironment provides a sensitive measure of disease progression in transgenic mice.