TREM2, Microglia and Neuroinflammation
An overview of TREM2, its role in microglia, links to neurodegenerative diseases, and potential treatment implications.
What is TREM2?
Triggering receptor expressed on myeloid cells 2 (TREM2) is a type-I transmembrane glycoprotein that serves as an innate immune receptor on microglia within the central nervous system (CNS) as well as on other myeloid cells (e.g. dendritic cells and granulocytes). This protein is encoded by the TREM2 gene and belongs to the TREM superfamily, which also includes TREM1 (Gratuze, 2018; Li, 2023). TREM2 plays a vital role in preserving brain homeostasis and responding to various pathological conditions (Qin, 2021; Li, 2023; Shi, 2025).
Signaling cascade:
- TREM2 functions as a sensor for a diverse range of ligands, including components derived from bacteria, phospholipids, glycolipids, APOE, APOJ, amyloid-beta oligomers, and TDP-43, among others (Filipello, 2022; Tagliatti, 2024; Shi, 2025).
- TREM2 interacts with the adaptor proteins DAP12 and DAP10, resulting in phosphorylation facilitated by the immunoreceptor tyrosine-based activation motif (ITAM) (Ulland, 2018; Li, 2023; Shi, 2025).
- This interaction triggers intracellular events, including the activation of spleen tyrosine kinase (SYK) and various downstream pathways, such as PI3K/AKT, mTOR, MAPK, and NF-κB (Ulland, 2018; Li, 2023; Shi, 2025).
- Soluble TREM2 (sTREM2) can also be derived from the proteolytic cleavage of TREM2's ectodomain by α-secretases (ADAM10, ADAM17), which releases the soluble ectodomain into the extracellular space, including the cerebrospinal fluid (CSF) and plasma. Additionally, sTREM2 can be produced directly through alternative splicing of the TREM2 mRNA (Ulland, 2018; Filipello, 2022; Hou, 2022; Tagliatti, 2024). sTREM2 is regarded as a biomarker for microglial activation and neuroinflammation in the brain (Filipello, 2022; Li, 2023).
Through the signaling cascade, the main functions of TREM2 include:
- Sensing damage: TREM2 ligands are frequently linked to tissue injury, neuronal death, demyelination, and the presence of pathological protein aggregates (Filipello, 2022; Tagliatti, 2024; Shi, 2025).
- Microglial survival and proliferation: The ITAM/Syk pathway activates the PI3K/AKT/mTOR signalling pathway, which not only inhibits apoptotic pathways, but also stimulates cell growth signals (Wang, 2015; Ulland, 2018; Qin, 2021; Shi, 2025).
- Phagocytosis and debris clearance: TREM2 mediates mobilization of calcium, alongside activation of diacylglycerol (DAG) and protein kinase C (PKC), leading to cytoskeletal rearrangement that is key for phagocytosis. Ultimately, TREM2 promotes the engulfment and clearance of cellular debris, including apoptotic neurons, myelin debris, and misfolded proteins, such as amyloid-beta aggregates in AD, and TDP-43 in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (Lue, 2015; Gratuze, 2018; Qin, 2021; Li, 2023; Shi, 2025). Please refer to our Resource titled "Microglia-Neuron Interactions & Neurodegenerative Diseases" for more information about phagocytosis in neurodegenerative conditions.
- Inflammatory regulation: TREM2 generally acts as a negative regulator of pro-inflammatory responses in microglia through the PI3K/AKT/Fox03a and PI3K/AKT/GSK3b pathways, promoting the secretion of IL-10 and TGF-β while inhibiting pro-inflammatory cytokines (TNF-α, IL-1β). However, it can also stimulate the production of pro-inflammatory cytokines and enhance phagocytosis via NF-κB and MAPK pathways (Gratuze, 2018; Li, 2023; Shi, 2025).
- Modulation of disease-associated microglia (DAM): TREM2 plays a crucial role in the transition of microglia from a homeostatic state to a DAM phenotype. This DAM state is thought to offer protective effects, mitigating neurodegeneration in conditions such as Alzheimer’s disease (AD) (Filipello, 2022; Basha, 2023; Li, 2023; Tagliatti, 2024).
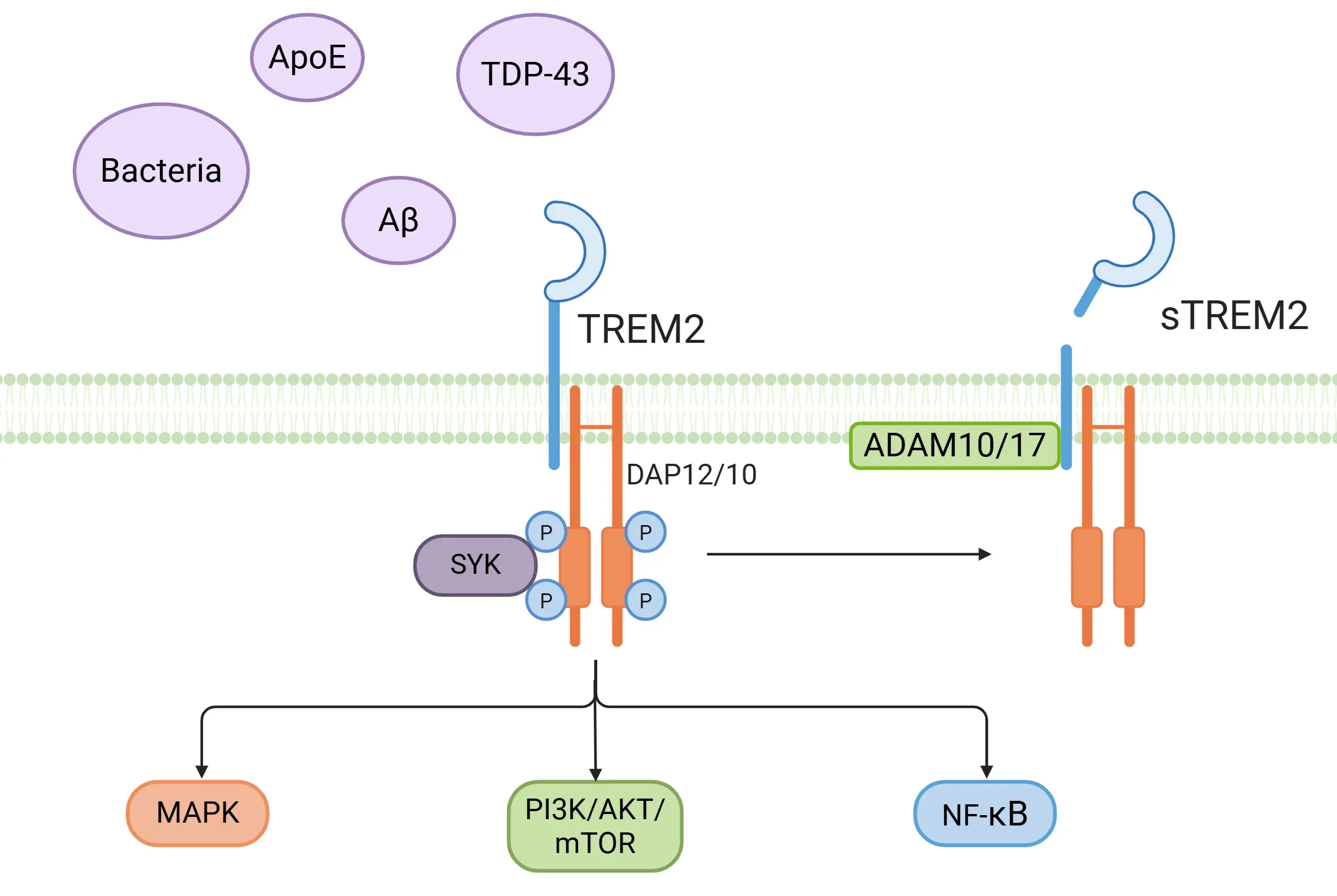
TREM2 signaling cascade.
Activation of the TREM2 receptor by various ligands triggers different signaling pathways and the release of soluble TREM2 (sTREM2).
What is the role of TREM2 in neurodegenerative diseases?
TREM2 plays a crucial role in the functioning of microglia, which are essential for maintaining brain homeostasis and have significant implications in neurological disorders. While TREM2 generally promotes protective mechanisms, dysregulation of this molecule can lead to increased inflammation and contribute to neurodegenerative conditions.
A biallelic mutation in the TREM2 gene results in a rare autosomal recessive disorder known as Nasu-Hakola disease. This condition leads to polycystic lipomembranous osteodysplasia, accompanied by sclerosing leukoencephalopathy. As a result, individuals often experience early-onset dementia (Carmona, 2018; Kiianitsa, 2024).
Heterozygous variants of the TREM2 gene mutation increase the risk of developing late-onset AD by reducing microglia activation and dysregulating the inflammatory response. Among neurodegenerative diseases, the role of TREM2 in AD is the most extensively studied. Below is an overview of the involvement of TREM2 in AD:
- TREM2 interacts with ligands such as phospholipids, ApoE-containing lipoproteins, or Aβ oligomers, binding to the adaptor DAP12. This interaction activates downstream signaling cascades that promote microglial survival, proliferation, chemotaxis, and the phagocytosis of apoptotic neurons and debris (Zhang, 2025).
- The TREM2 signaling cascade also facilitates the transition of homeostatic microglia into DAM that cluster around amyloid plaques. These DAMs compact the plaques and form a protective barrier that limits neuritic dystrophy, particularly in the early stages of AD (Zhang, 2025).
- In mouse models, TREM2 deficiency results in fewer DAM clustering around amyloid-beta plaques, leading to more diffuse plaques, an elevated Aβ42/Aβ40 ratio, and increased axonal damage. Conversely, TREM2 overexpression or treatment with agonistic antibodies can restore microglial responses, enhance plaque clearance, and improve glucose metabolism (Ulland, 2018; Zhang, 2025).
- The role of TREM2 in tau pathology is complex, showing mixed results. Its effect on limiting tau accumulation and propagation is limited to the presence of amyloid-beta plaques (Zhang, 2025).
- Loss-of-function variants (such as R47H, R62H, and H157Y) reduce ligand affinity or increase ectodomain shedding, thereby impairing microglial activation and cell metabolism. This exacerbates both amyloid and tau pathology and raises the risk for AD (Guerreiro, 2013; Gratuze, 2018; Zhang, 2025).
- sTREM2 generated by ADAM-mediated shedding appears in CSF early in AD. sTREM2 correlates with microglial activation and slower cognitive decline, and it can bind Aβ oligomers to inhibit aggregation, making it a promising diagnostic biomarker and therapeutic target (Zhang, 2025).
TREM2 appears to be involved in neurodegenerative diseases beyond AD, although its exact role remains less defined (Carmona, 2018; Kiianitsa, 2024; Awuah, 2025):
- Rare heterozygous TREM2 variations have been linked to an increased risk of FTD, while the p.Arg47His variant has been observed in Parkinson's disease (PD) cohorts. However, associations in these conditions remain inconsistent and vary by population (Carmona, 2018; Huang, 2023).
- There are studies investigating the relationship between TREM2 variations and the risk of conditions such as ALS, Lewy body dementia, Creutzfeldt-Jakob disease, and ischemic stroke, among others. Nonetheless, these association studies have produced mixed results, and the mechanisms by which TREM2 influences these diseases remain to be elucidated (Carmona, 2018; Awuah, 2025).
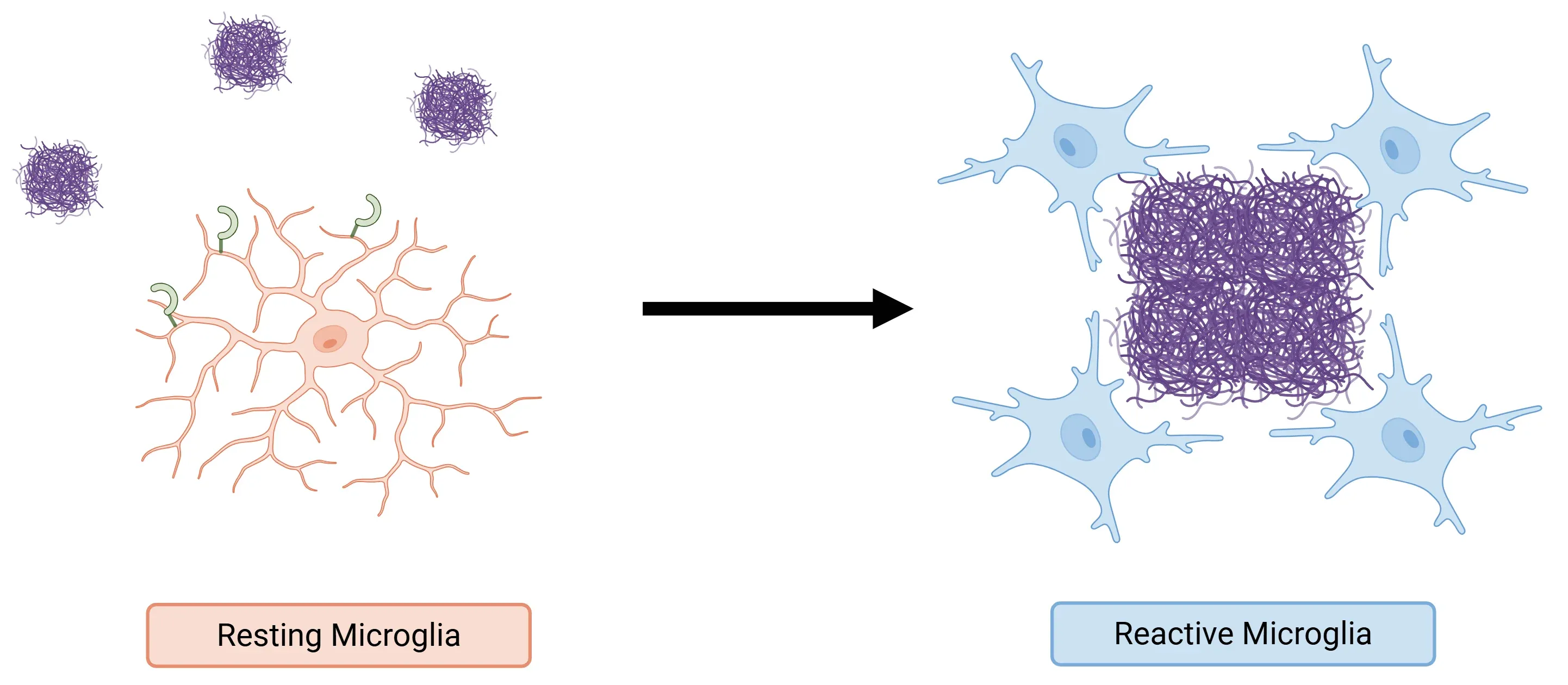
Role of TREM2 in AD.
By binding to amyloid-beta, TREM2 activates microglia, transforming them into DAM. These cells cluster around amyloid plaques to mitigate the damage caused by these plaques in the initial stages of the disease.
Is TREM2 a potential therapeutic target?
TREM2 is a promising therapeutic target for AD and possibly for other neurodegenerative disorders. Mechanistically, TREM2 signalling promotes microglial survival, proliferation, lipid metabolism, and phagocytosis of amyloid‑β and debris, thereby limiting plaque formation and neuritic dystrophy.
Preclinical research supports the therapeutic potential of TREM2:
- TREM2 agonist antibodies, including 4D9 and AL002, have been shown to increase microglial proliferation, improve plaque compaction, and enhance brain glucose metabolism in transgenic mouse models of AD (van Lengerich, 2023; Awuah, 2025).
- The ATV:TREM2 antibody, which crosses the blood-brain barrier (BBB) using a transferrin receptor binding site, enhances microglial activity and glucose metabolism in AD mouse models (van Lengerich, 2023).
- 5xFAD mouse models that overexpress the TREM2 gene have shown improved phagocytosis of amyloid-beta and enhanced cognitive function (Li, 2023).
- While the specific role of soluble TREM2 (sTREM2) remains unclear, administering sTREM2 has been found to reduce neuritic dystrophy in amyloidogenic mice. However, alterations in TREM2 shedding in transgenic mice can lead to increased amyloid deposits (Zhang, 2025).
Clinical translation of these findings is already in progress:
- The humanized antibody AL002 has completed Phase 1 safety trials, demonstrating favorable tolerability without serious adverse events (Wang, 2015; Long, 2024). However, the outcomes of the Phase 2 trials have not yielded positive results.
- VHB937 is a TREM2 agonist monoclonal antibody that has been shown to enhance microglial phagocytosis and chemotaxis. This therapeutic agent is currently undergoing evaluation in Phase 2 clinical trials for the treatment of ALS (Noh, 2025).
- TREM2 has a complex role; its activation appears most beneficial during the early stages of amyloid seeding. In later stages of the disease, different strategies may be required, and off-target effects on peripheral myeloid cells must also be considered (Zhang, 2025).
In summary, TREM2 shows significant promise as a therapeutic target for AD and possibly other neurodegenerative disorders. Preclinical studies emphasize its role in enhancing microglial function and improving cognitive outcomes. However, due to TREM2’s intricate role in disease progression, careful consideration is necessary. Ongoing research is critical to optimizing TREM2-targeted therapies for effective intervention in neurodegeneration.
Our team would be happy to answer any questions about TREM2 and Microglia or provide specific information about the neurodegenerative disease models we use for therapeutic efficacy studies.
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
APOE4, Microglia & Alzheimer’s Disease
An overview of how ApoE4 influences microglial activity in Alzheimer's disease and the development of targeted therapeutics.
Microglia-Neuron Interactions & Neurodegenerative Diseases
A concise review of the direct interactions between microglia & neurons, and how these cell-to-cell interactions may be affected in neurodegenerative diseases.
TNF-α (TNF-alpha) & Microglia in Neurodegenerative Diseases
An overview of the function of tumor necrosis factor-alpha (TNF-α) in microglia and its contribution to the progression of neurodegeneration.
What Is IL-1β (IL-1b)? Function, Signaling, and Biological Role
An overview of IL-1β, including its signaling pathways, involvement in disease mechanisms, and potential therapeutic targets.
Amyloid-β Plaque Analysis in Alzheimer's Disease
Overview of methods to classify & quantify Aβ plaques in brain tissue sections from humans & Alzheimer’s disease animal models (transgenic mice & rats).
Microglia Morphology in ALS, Alzheimer's Disease & Parkinson's Disease
An overview of microglial morphological analysis and the applications to neurodegenerative disease research and drug discovery & development.
Microglial Activation in an α-Synuclein PFF Mouse Model
We have quantified microglial activation, based on morphology, in an α-synuclein preformed fibril (PFF) seeding & spreading mouse model of Parkinson’s disease.
Amyloid-β & Inflammatory Microenvironment in Alzheimer's Mice
We have analyzed the complex spatial relationships between β-amyloid plaques, activated & resting microglia, and astrocytes in an APP/PS1 transgenic model.