Mitochondrial Dysfunction & Parkinson's Disease
An overview of how mitochondrial dysfunction is associated with neurodegeneration in Parkinson’s disease.
What is mitochondrial dysfunction?
Mitochondria are double-membrane organelles that play a central role in managing cells’ metabolic activities through the production of adenosine triphosphate (ATP), the primary energy currency that powers cellular functions (Casanova, 2023; Chen, 2023). The inner mitochondrial membrane houses an array of proteins essential for electron transport and ATP synthesis through oxidative phosphorylation, the process of releasing chemical energy that gets converted into ATP. The outer mitochondrial membrane (OMM), on the other hand, is permeable to small molecules and acts as a sieve, allowing the passage of metabolites required for oxidative phosphorylation. Beyond their role as energy factories, these highly dynamic organelles are also involved in other essential functions, such as regulating calcium homeostasis, epigenetic signaling, metabolic signaling, and programmed cell death (Bhatti, 2017; Collier, 2023; Nyugen, 2023).
The origins of all mitochondria can be traced back to a common ancestral organelle, as mitochondria cannot be formed de novo (Shiota, 2015; Roger, 2017). Instead, they exist within a complex interconnected system, where they divide independently of the cell cycle through fisson, a process that helps increase energy production in response to rising cellular energy demands. Conversely, mitochondrial fusion allows two smaller mitochondria to form a larger mitochondrion, promoting the exchange of resources and other components between mitochondria for optimal functioning. Hence, maintaining a healthy mitochondrial population requires the selective removal of damaged and dysfunctional mitochondria from the mitochondrial network through mitophagy, a type of selective autophagy, that tags, degrades, and recycles dysfunctional mitochondria (Ashrafi, 2013; Uoselis, 2023).
Mitochondrial dysfunction, characterized by impaired mitochondrial function and an inability to satisfy cellular energy demands, is implicated in the pathogenesis of several neurodegenerative diseases, including Parkinson’s disease (PD), Alzheimer’s disease (AD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) (Beal, 2000; Johri, 2012; Norat, 2020). Neurons, due to their high energetic demands and reliance on mitochondria for calcium buffering, are particularly vulnerable to impaired mitochondrial function (Paß, 2021; Henrich, 2023).
Several factors contribute to and exacerbate mitochondrial dysfunction in neurodegenerative diseases, including:
- Reduced or excessive mitophagy, which results in an inadequate mitochondrial population
- Disruptions in the availability or transport of substrates required for ATP production due to defects in the electron transport chain and ATP-synthesis machinery
- Excessive production of reactive oxygen species (ROS), which amplifies mitochondrial damage and induces excessive mitophagy
- Impaired calcium regulation, which leads to disruptions in cellular signaling
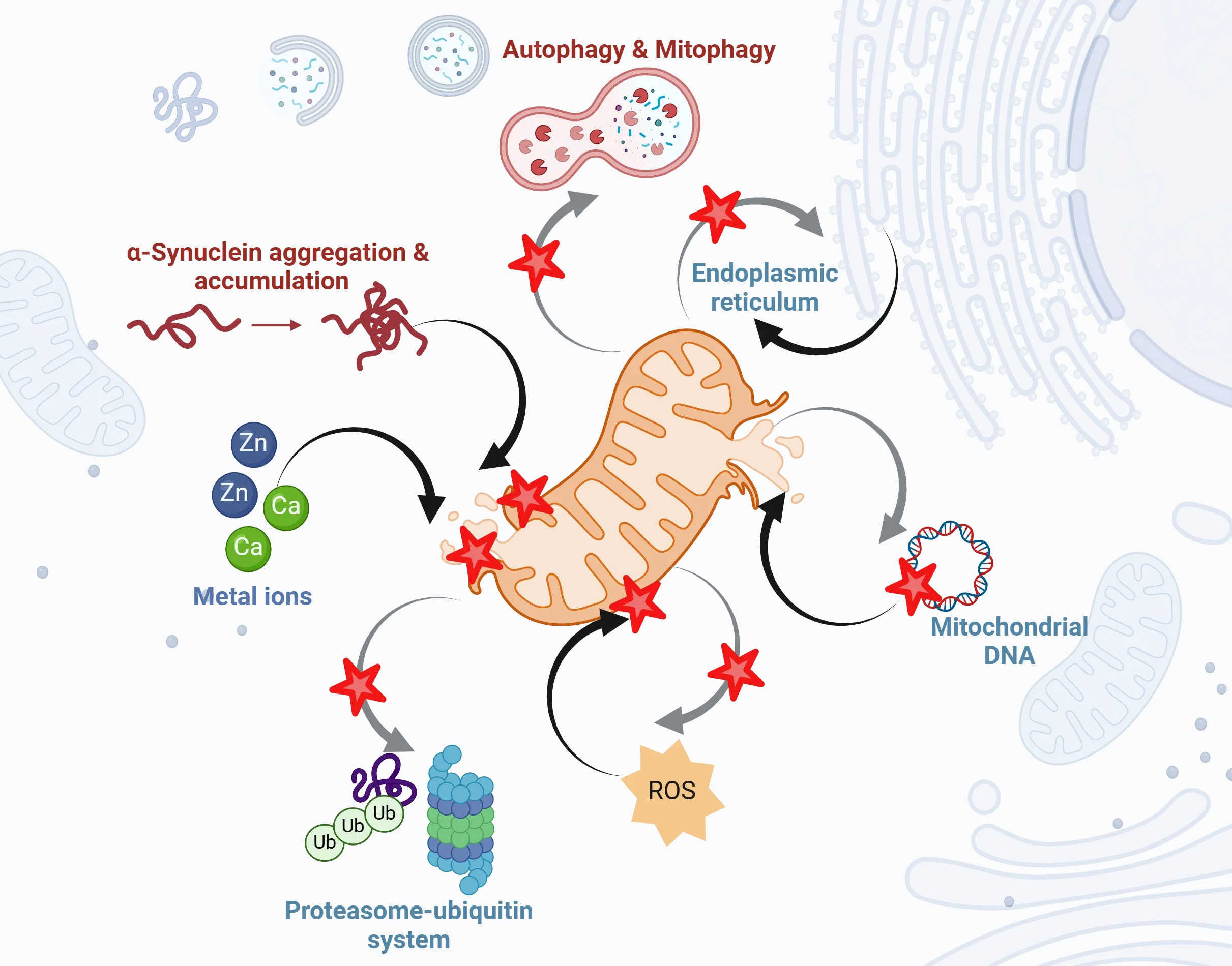
Mitochondrial dysfunction plays a central, yet complex role in the pathogenesis of Parkinson's disease. The schematic diagram highlights common mechanisms of mitochondrial dysfunction with black arrows depicting direct and indirect influences, such as protein aggregation, Zn and Ca imbalance, ROS, and mtDNA damage. Figure and caption are reproduced from Prasuhn et al. (Prasuhn, 2021) under the Creative Commons Attribution License.
What is the relationship between mitochondrial dysfunction and degeneration of dopaminergic neurons in Parkinson's Disease?
The degeneration of dopaminergic neurons in PD is closely linked to mitochondrial dysfunction. Due to their exceptionally high energetic demands, these neurons rely on a vast and complex mitochondrial network that extends from dendrites to synaptic terminals. Mitochondrial oxidative phosphorylation, essential for ATP generation, is impaired in mitochondrial dysfunction. However, a continuous ATP supply is necessary for maintaining ionic homeostasis, driving synaptic activity and transmitter recycling—processes that support the function and survival of dopaminergic neurons (Sheng, 2017; Duarte, 2023; Henrich, 2023). In addition, mitochondria also play an essential role in intracellular calcium buffering, which is essential for proper axonal function (Walters, 2023). Hence, disruptions in the aforementioned mitochondrial processes adversely impact neuronal health and are key contributors to PD pathology.
Mitochondrial dysfunction in PD is further exacerbated by alpha-synuclein pathology, a hallmark of PD (Geibl, 2024). Certain alpha-synuclein species, for instance, bind to the outer mitochondrial membrane protein TOM20, preventing its interaction with its co-receptor TOM22 and impairing mitochondrial protein import (Di Maio, 2016). Overexpression of alpha-synuclein also induces the opening of the mitochondrial permeability transition pore (mPTP), a protein channel that maintains mitochondrial integrity (Parihar, 2008; Ludtmann, 2018). This opening leads to the loss of mitochondrial membrane potential, reduced ATP production, release of harmful reactive oxygen species (ROS), and cell death.
Mutations in genes encoding mitochondrial quality control proteins are also strongly associated with degeneration of dopaminergic neurons (de Castro, 2011). For example, Parkin knockout mice exhibit reduced mitochondrial respiratory capacity, while PINK1 knockout mice experience deficits in calcium buffering capacity and impairments in mitochondrial membrane potential (Palacino, 2004; Akundi, 2011). Genetic mutations in lysosomal function-related proteins, such as GBA1, LRRK2, VPS35, also contribute to neuronal loss. Studies in DAT-Cre mice have revealed that loss-of-function mutations in these genes disrupt mitochondrial dynamics, leading to in PD-like neurodegeneration (Tang, 2015).
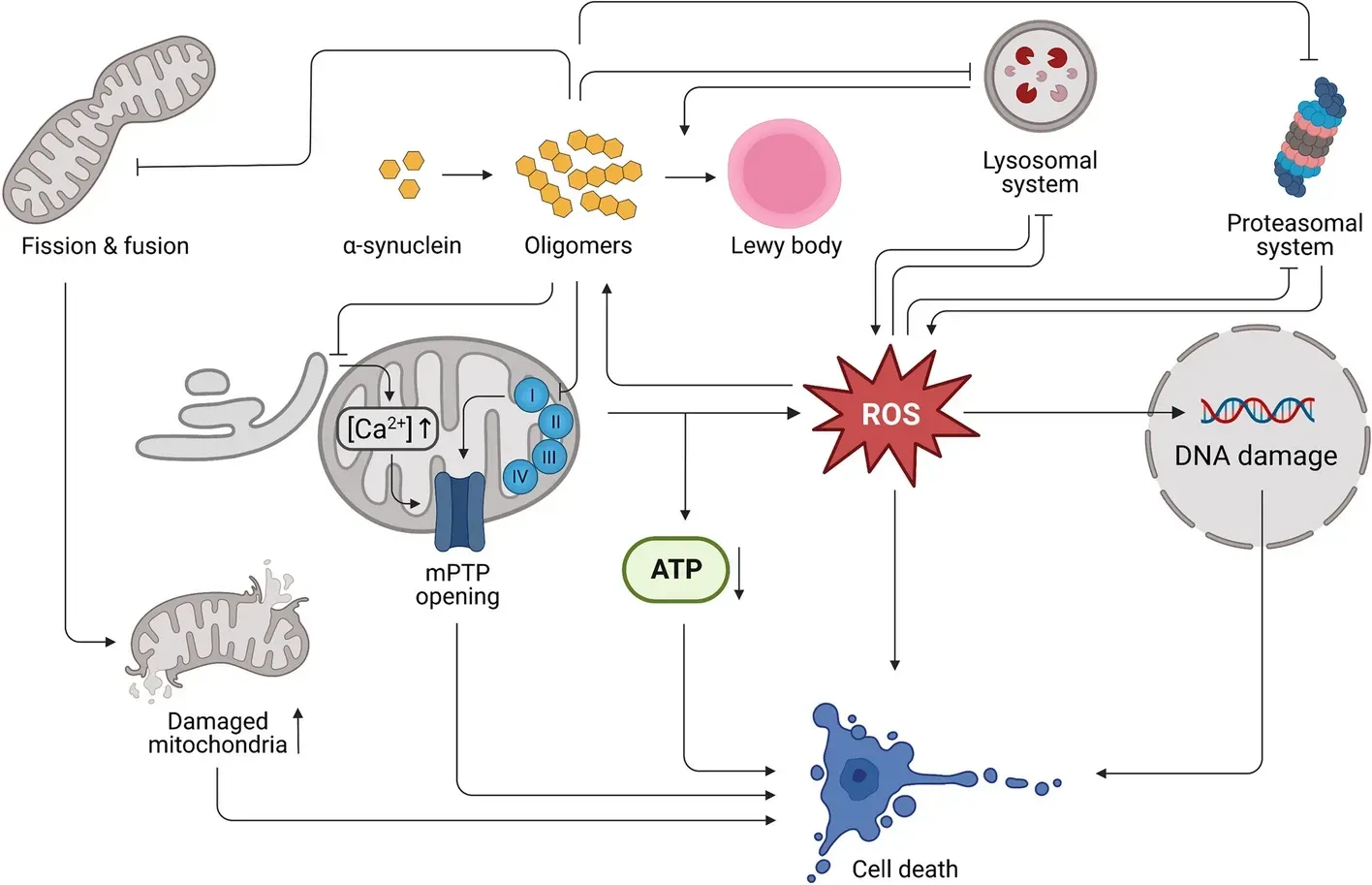
Mitochondrial dysfunction in PD is exacerbated by alpha-synuclein pathology. The diagram describes several mechanisms of alpha-synuclein induced cellular dysfunction and death. Figure is reproduced from Henrich et al. (Henrich, 2023) under the Creative Commons Attribution License.
Are there any therapeutic approaches targeting mitochondrial dysfunction in Parkinson's Disease?
Given the close association between mitochondrial dysfunction and neurodegeneration, several therapeutic approaches are currently under investigation to mitigate mitochondrial damage in PD. These strategies include:
Mitophagy Enhancers
These are small molecules that either directly activate the critical mitochondrial quality control proteins, PINK1 and Parkin, or inhibit the function of Parkin’s counterplayer, the ubiquitin-specific protease USP30. Some mitophagy enhancers currently under investigation for their therapeutic potential are urolithin A, nicotinamide riboside, actinonin, tomatidine, and spermidine. In preclinical models of neurodegeneration, these compounds have been shown to enhance mitophagy by facilitating the clearance of defective and dysfunctional mitochondria from affected cells, which results in improved synaptic and cognitive function (Miller, 2019; Kshirsagar, 2021; Wang, 2022). Additionally, FDA-approved drugs, such as rapamycin and metformin, are also being studied for their potential to enhance mitophagy (Sarkar, 2007). Rapamycin acts by inhibiting the mTOR signaling pathway, which is a negative regulator of autophagy. Metformin, on the other hand, activates the AMPK signaling pathway, which triggers PINK1/Parkin dependant mitophagy (Bharath, 2020).
Mitochondrial Permeability Transition Pore (mPTP) Inhibitors
Opening of the mPTP is a key driver of cell death in neurodegenerative diseases (Bauer, 2020). Cyclophilin D (CypD) is the most crucial molecule for mPTP formation, and acts as a regulatory protein in cell death. Small molecule inhibitors targeting CypD activity have emerged as an attractive therapeutic strategy for preventing permeability transition, mitigating mitochondrial dysfunction, and slowing down neuronal loss in PD. One CypD inhibitor currently being investigated for its therapeutic potential in preclinical models of neurodegeneration is cyclosporin A (CsA) (Samanta, 2024).
Mitochondrially-targeted Antioxidants
Mitochondrial dysfunction is a key driver of ROS. Their overproduction can lead to heightened oxidative stress, damage to mitochondrial DNA, disruptions in neuronal energy metabolism, and reduced cell viability. These factors play a critical role in the progression of neurodegenerative diseases (Andersen, 2004; Szeto, 2006; Dash, 2024).
In order to combat the harmful effects of ROS, scientists are developing mitochondrially-targeted antioxidants to selectively accumulate within dysfunctional mitochondria, where they can neutralize ROS at the source, prevent mitochondrial oxidative damage, and restore energy supply to neurons (Jin, 2014; Apostolova, 2015). Some mitochondrially-targeted antioxidants currently being investigated for their activity and therapeutic potential include:
- Mitoquinone Mesylate (MitoQ)
A derivative of ubiquinone conjugated to triphenylphosphonium. MitoQ enters the mitochondrial matrix, scavenges free radicals, and lowers their concentration (Duarte, 2023). - Mitochondria-targeted Vitamin E (MitoVitE)
The chromanol moiety of vitamin E bound to a triphenylphosphonium cation. It penetrates through lipid bilayers, accumulating within mitochondria (Smith, 1999). - Mitochondria-targeted Apocynin (MitoApocynin)
A triphenylphosphonium conjugated derivative of apocynin. It inhibits the production of superoxide radicals, which are a major cause of cellular oxidative stress (Ghosh, 2016).
Gene Therapy
Loss of function mutations in genes encoding mitochondrial quality control proteins PINK1 and Parkin can lead to impaired mitophagy and insufficient removal of dysfunctional mitochondria. Hence, gene augmentation therapies that boost the PINK1/Parkin pathway have the potential to enhance mitophagy and mitigate mitochondrial dysfunction (Quinn, 2020).
Our team would be happy to answer any questions about mitochondrial dysfunction in neurodegenerative diseases or provide specific information about the PD models we use for therapeutic efficacy studies.
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on mitochondrial dysfunction and Parkinson's disease in neurodegenerative diseases and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
Mitochondrial Dysfunction in Microglia & Astrocytes
The role of mitochondrial dysfunction in microglia and astrocytes in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and ALS.
Mitophagy and Parkinson’s Disease
An overview of how impaired mitophagy can lead to neurodegeneration in Parkinson’s disease.
Autophagy, Parkinson's Disease, and Dopaminergic Neurons
An overview of how impaired autophagy can lead to pathologic changes and neurodegeneration in dopaminergic neurons in Parkinson’s disease.
Autophagy & Neurodegenerative Diseases
An overview of how cellular autophagy plays a role in brain health and neurodegeneration.