A Guide to Mouse Models of Frontotemporal Dementia (FTD)
A Resource providing a comprehensive overview of animal models used in frontotemporal dementia research, including genetic and pathological disease mechanisms.
Table of Contents
What is frontotemporal dementia (FTD)?
Clinical Features and Subtypes
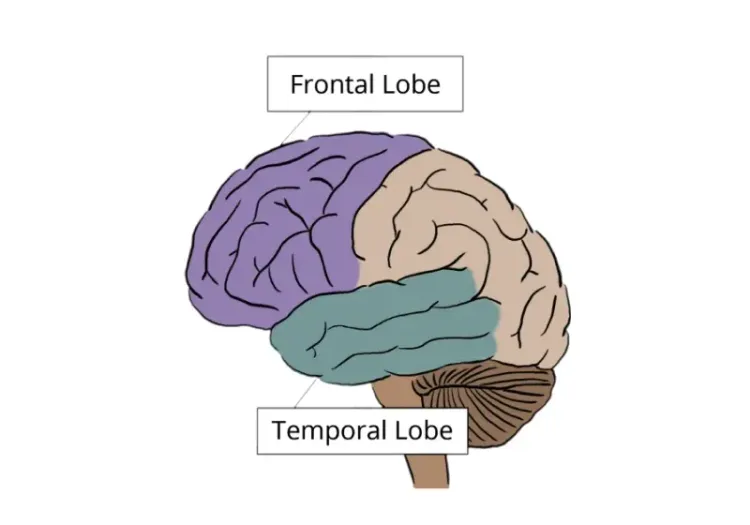
Frontotemporal Dementia (FTD) is a common type of dementia, particularly affecting individuals under the age of 65. Alzheimer’s disease (AD) is the most common cause of early-onset dementia, followed by vascular dementia and frontotemporal lobar degeneration (FTLD) (Vieira, 2013). FTD comprises a clinically and pathologically heterogeneous group of neurodegenerative syndromes associated with progressive degeneration of the frontal and temporal lobes. Clinically, FTD represents the symptomatic manifestation of underlying FTLD pathology and is characterized by progressive impairments in behavior, executive function, and/or language (Root, 2021).
FTD is broadly classified into distinct clinical syndromes based on the predominant symptoms at onset. The most common presentation is the behavioral variant of FTD (bvFTD), accounting for approximately 60–80% of cases. The behavioral variant of FTD is characterized by early and progressive changes in personality and social conduct, including behavioral disinhibition, apathy, compulsive or stereotyped behaviors, and a loss of insight. As the disease progresses, patients may also develop language impairments, Parkinsonian features, or motor neuron disease (MND).
Two major language variants of FTD are classified under the umbrella of primary progressive aphasia (PPA):
Semantic variant PPA (svPPA) is characterized by progressive loss of semantic knowledge, including impaired word comprehension, and diminished recognition of objects and faces. Patients typically present with fluent but empty speech and severe anomia.
Non-fluent/agrammatic variant PPA (nfvPPA) is defined by non-fluent, effortful speech. Clinical features may include agrammatism in spoken and written language and apraxia of speech—a motor planning disorder that disrupts word production (Whitwell, 2019).
Despite advances in our understanding of the clinical spectrum and genetic underpinnings of FTD, there are currently no approved disease-modifying therapies. Existing treatments are symptomatic and aim to manage behavioral or cognitive symptoms, rather than alter disease progression, underscoring the need for robust preclinical models.
Neuropathology and Genetics
FTD is neuropathologically heterogeneous and is most commonly defined by the abnormal aggregation of one of three proteins: microtubule-associated protein tau, TAR DNA-binding protein 43 (TDP-43), or fused in sarcoma (FUS). Based on the predominant pathological protein, FTD is subclassified into FTLD-tau, FTLD-TDP, and FTLD-FUS.
Importantly, clinical presentation does not reliably predict the underlying molecular pathology. For example, bvFTD may be associated with any of the three major pathological forms. In contrast, svPPA is most consistently linked to FTLD-TDP, while nfvPPA is predominantly associated with FTLD-tau pathology.
A substantial proportion of FTD cases are familial, typically following an autosomal dominant inheritance pattern. The most frequently implicated genes include MAPT, GRN (progranulin), and a GGGGCC hexanucleotide repeat expansion in C9orf72. While MAPT mutations are associated with FTLD-tau, both GRN and C9orf72 mutations are linked to FTLD-TDP pathology. All three mutations commonly present as bvFTD, although GRN and C9orf72 carriers often exhibit broader phenotypic variability, including language deficits, Parkinsonism, or amyotrophic lateral sclerosis (ALS) (Whitwell, 2019).
To discover more about FTD’s neuropathological features, visit our Resource: Neuroimaging in Frontotemporal Dementia and Clinical Trials.
It is now widely recognized that FTD and ALS exist as a continuum of neurodegenerative disease, sharing overlapping genetic, clinical, and pathological features. The most prominent genetic link between the two conditions is the C9orf72 repeat expansion with less frequent mutations in TARDBP, SQSTM1, UBQLN2, CHMP2B, CHCHD10, and VCP, further supporting a shared pathogenic spectrum (Lopez-Herdoiza, 2023; Genin, 2024).
Learn more about ALS in our Resource: A Guide to ALS Models for Drug Discovery.
Why are mice used as animal models of FTD?
Given the clinical and pathological heterogeneity of FTD, a wide range of transgenic and AAV-mediated mouse models have been developed to recapitulate key molecular and neuropathological features of the disease. Mice remain the most widely used species in FTD research due to their genetic tractability, conserved neuronal circuitry, and suitability for detailed molecular, cellular, and functional investigations. Most FTD mouse lines are engineered to express disease-associated mutations or proteins, enabling mechanistic studies at the gene and protein level relevant to FTD pathogenesis (Ahmed, 2017).
FTD primarily affects the salience network, which includes the anterior insula, anterior cingulate cortex, ventral striatum, amygdala, dorsomedial thalamus, hypothalamus, and several brainstem nuclei, including the periaqueductal gray, substantia nigra, and ventral tegmental area. These interconnected regions govern fundamental physiological and behavioral processes such as pain, hunger, thirst, autonomic regulation, reward processing, motivation, and fear responses. Importantly, the core architecture and connectivity of this network are highly conserved across species, supporting the use of mice to model key aspects of FTD-related circuit dysfunction (Roberson, 2012).
Behavioral Domains Affected in FTD and Their Relevance to Mouse Models
Several behavioral domains disrupted in FTD rely on conserved neuroanatomical circuits and can therefore be partially modeled in rodents using validated behavioral paradigms (Kesner, 2011; Bizon, 2012; Roberson, 2012; Hamilton, 2015; Mora, 2023):
Social Dysfunction: Reduced social engagement and loss of empathy in FTD are linked to medial prefrontal cortex (PFC) dysfunction. Comparable deficits are observed in mutant mice with dorsomedial PFC involvement. The anterior cingulate cortex—also implicated in FTD—plays a key role in modulating empathic and social behaviors in rodents.
Repetitive Behaviors: Pathological grooming and stereotyped behaviors observed in certain knockout mouse models parallel compulsive and repetitive behaviors seen in FTD. The striatum, implicated in both species, plays a central role in these behaviors.
Emotional Dysregulation: FTD patients often show blunted responses to negative emotions, associated with amygdala atrophy. Similarly, mice with disrupted amygdala function exhibit impaired fear conditioning, supporting the translational value of these models.
Executive Deficits: Progressive impairment in planning, decision-making, and working memory are core features of FTD and involve dysfunction of the dorsolateral PFC, anterior cingulate cortex, medial PFC, and basal ganglia. In FTD mouse models, disruption of the PFC– striatal–thalamic circuitry similarly leads to executive dysfunction. These deficits are commonly assessed using tasks such as the T-maze, Y-maze, and attentional set-shifting paradigms.
Cognitive Flexibility: Difficulty shifting attention or behavior and rigid thinking in FTD are linked to the orbitofrontal cortex (OFC), PFC, mediodorsal thalamus, and basal ganglia. OFC atrophy is associated with behavioral inflexibility in both FTD patients and rodent models. Reversal learning and touchscreen operant shift tests are used to measure cognitive inflexibility.
Motor Dysfunction: Motor symptoms occur especially in FTD-ALS spectrum or FTD with tau mutations involving the motor cortex (primary and supplementary motor area), basal ganglia, and brainstem motor pathways. In FTD rodent models, motor phenotypes are linked to cortical and basal ganglia degeneration. Rotarod performance, grip strength, gait analysis, and clasping are commonly used to quantify motor decline.
To learn more about behavioral and motor assays commonly used to track disease progression, see: Motor and Sensory Function Tests and Sleep and Cognition Tests.
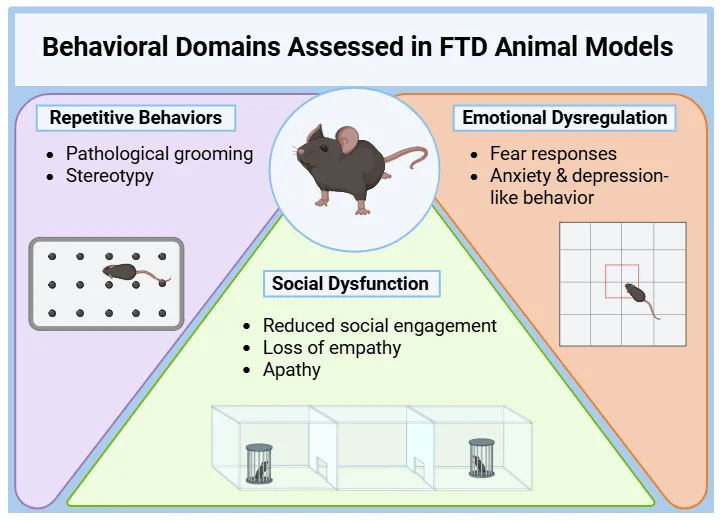
Comprehensive behavioral assessments of FTD mouse models, including measures of repetitive behaviors, social interaction, and emotional reactivity, provide robust endpoints for the preclinical evaluation of disease-modifying therapies.
Important Considerations for Mouse Models in FTD Research
Mouse models of FTD have primarily been designed to replicate the core behavioral features of bvFTD, including social withdrawal, disinhibition, compulsive behaviors, and stereotyped motor patterns. This focus is both practical and clinically relevant, as bvFTD is the most prevalent FTD subtype, occurring approximately four times more frequently than PPAs (Hogan, 2016).
These preclinical models have proven invaluable for uncovering disease mechanisms, mapping affected neural circuits, and identifying potential therapeutic targets within accessible, well-characterized systems. However, modeling language-predominant forms of FTD— svPPA and nfvPPA—remains a major challenge. These syndromes rely on complex linguistic and semantic networks that do not have direct homologs in the rodent brain, making it infeasible to fully recapitulate language-related deficits in mice.
Consequently, preclinical research has appropriately emphasized behavioral dimensions that are both robustly impaired in FTD and experimentally tractable in animal models. To address the limitations of rodent systems, researchers increasingly integrate complementary human-based approaches, such as induced pluripotent stem cell (iPSC)-derived neurons, brain organoids, and patient neuroimaging studies, to investigate aspects of language and semantic network pathology (Roberson, 2012; Whitwell, 2019).
Despite these constraints, mouse models remain an essential component of the translational research pipeline. They provide critical insights into early cellular and circuit-level mechanisms of FTD and offer a robust platform for evaluating candidate therapeutics in vivo.
What are the main FTD mouse models used in preclinical research?
Tau-Based Models (MAPT)
Mutations in the tau gene (MAPT) are a major genetic cause of FTD, alongside mutations in GRN and C9orf72. More than 40 FTD-associated MAPT mutations have been identified, most of which are missense mutations within the microtubule-binding region. These mutations produce both loss-of-function effects, by reducing microtubule stabilization, and gain-of-function effects, through enhanced tau aggregation and hyperphosphorylation (Roberson, 2012).
Mutant forms of human tau, including P301S and P301L, are associated with tauopathies like FTD with parkinsonism-17 (FTDP-17).
At Biospective, we generate Tau mouse models using multiple approaches:
Tau Pre-Formed Fibrils (PFF) Seeding: Enables propagation of pathological tau in vivo, facilitating studies of tau spreading and aggregation. Explore the model here: Tau Fibil Spreading Models.
AAV-hTau Delivery: Intracranial injection of AAV vectors into the brains of adult C57BL/6 mice recapitulates key human tauopathy features, including phosphorylated tau aggregates, behavioral dysfunction, and brain atrophy detectable by in vivo MRI. This approach allows for generation of readouts in a relatively short timeframe. View our Interactive Image Presentation here: AAV Tau Mouse Models.
Key Tau Mouse Models
rTg(TauP301L)4510
This model expresses the human P301L tau mutation in the forebrain, including the hippocampus and neocortex, with transgene expression that is regulatable using doxycycline. The mice exhibit age-dependent neurofibrillary tangle (NFT) formation, accompanied by pronounced forebrain atrophy and neuron loss.
Behavior: Mice display early hyperactivity and reduced anxiety-like behavior, followed by spatial memory deficits beginning around four months of age and progressive motor impairments, including a characteristic clasping reflex (Lewis, 2000; Ramsden, 2005; Pennanen, 2006).
AAV-TauP301L
In this model, human P301L tau is delivered intracranially using AAV vectors, leading to widespread tau expression in targeted brain regions. This approach results in tau hyperphosphorylation, the formation of pre-tangles and mature NFTs, dystrophic neurites, and neuroinflammatory responses.
Behavior: Mice exhibit hyperactivity, disinhibition, impaired fear conditioning, and memory deficits, reflecting dysfunction in hippocampal- and amygdala-dependent circuits (Cook, 2015; Silva-Llanes, 2025).
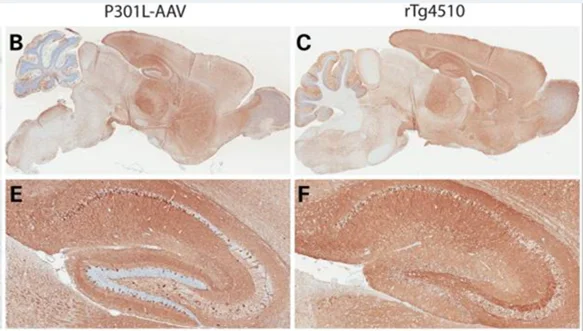.webp)
Widespread human tau expression in the AAV-TauP301L mouse model (panels B, E) closely mirrors the distribution observed in rTg4510 mice (panels C, F). Figure reproduced from Cook et al. (Cook, 2015) under the Creative Commons Attribution License.
P301S Tau
This transgenic model (e.g. PS19 mice) expresses the human P301S tau mutation and is characterized by early-onset behavioral and cognitive impairments that emerge prior to extensive NFT pathology. Synaptic dysfunction is observed as early as three months of age, followed by progressive tau accumulation, neuronal loss, and brain atrophy by nine to twelve months.
Behavior: Early phenotypes include hyperactivity and reduced anxiety-like behavior, with spatial memory deficits detected in the Morris water maze (Takeuchi, 2011).
Pro-Aggregation Tau (hTau40, ΔK280)
This model uses regulatable forebrain expression of a pro-aggregation variant of full-length human tau containing the ΔK280 mutation, which enhances β-structure formation and tau aggregation (Eckermann, 2007). Although no overt neuronal loss is observed, mice show marked synaptic deficits and reduced long-term potentiation.
Behavior: After prolonged gene expression, mice develop severe learning and memory impairments, while motor function remains intact (Van der Jeugd, 2012).
MAPT P301S;Int10+3;S320F Knock-In
This triple-mutant knock-in model displays robust and early tau pathology, progressive accumulation of pathological tau, synaptic loss, and severe atrophy in the hippocampus, hypothalamus, and amygdala, accompanied by astrogliosis.
Behavior: Mice exhibit increased anxiety, repetitive and stereotyped behaviors, impaired sustained attention, apathy-like behavior, and deficits in learning and behavioral flexibility (Morito, 2025).
TDP-43-Based Models
TDP-43-positive inclusions represent the most common pathological substrate in FTD (FTD-TDP). TDP-43 is a nuclear RNA- and DNA-binding protein involved in RNA processing and regulation. Pathogenic mutations in TARDPB lead to mislocalization of TDP-43 from the nucleus to the cytoplasm, resulting in a combination of loss of nuclear function and gain of toxic cytoplasmic function (Roberson, 2012).
Learn more about Biospective’s TDP-43ΔNLS (rNLS8) Mouse Model.
Key TDP-43 Mouse Models
TDP-43 Q331K Transgenic
This model expresses the human TARDBP Q331K mutation under the murine prion promoter, resulting in near-physiological expression levels and downregulation of endogenous mouse TDP-43. The protein remains predominantly nuclear and does not form classic cytoplasmic aggregates (Wong, 2020; Watkins, 2021).
Behavior: Mice develop tremors, abnormal gait, reduced muscle mass, and frontal cortex-mediated cognitive deficits, while hippocampus-dependent memory remains largely preserved (Wong, 2020; Watkins, 2021).
TDP-43 Q331K Knock-In
In this physiologically relevant model, the Q331K mutation is introduced into the endogenous mouse TARDBP gene, avoiding overexpression artifacts. The model recapitulates key structural brain changes seen in ALS-FTD, including atrophy of the frontal and entorhinal cortices and hippocampus, along with widespread microglial activation (White, 2018; Lin, 2021).
Behavior: Mice exhibit impairments in learning, attention, and memory (White, 2018; Lin, 2021).
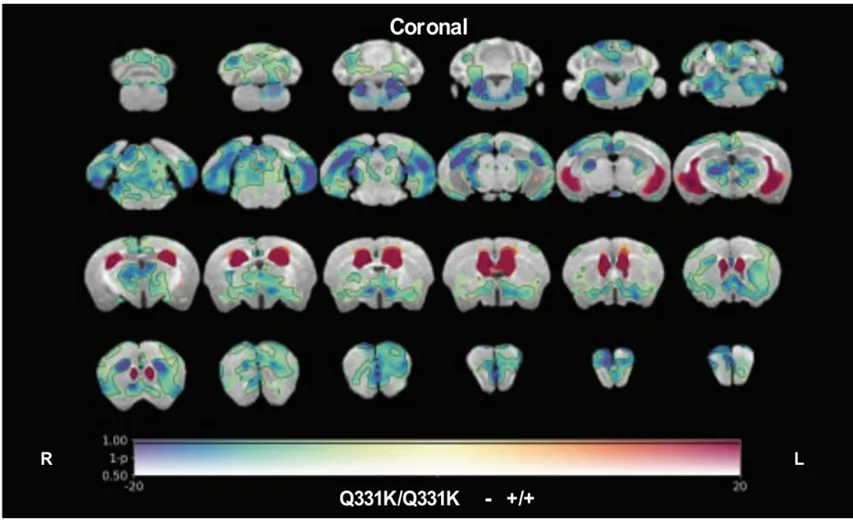.webp)
In vivo magnetic resonance imaging (MRI) reveals regional brain volume loss (cool colors) and ventricular enlargement (warm colors) in TDP-43Q331K/Q331K knock-in mice compared with age-matched wild-type controls, highlighting structural changes that mirror human ALS-FTD pathology. Figure reproduced from Lin et al. (Lin, 2021) under the Creative Commons Attribution Licence.
CamKIIa-hTDP-43NLSm Inoculation
This model uses transgenic mice expressing a cytoplasmically mislocalized form of human TDP-43 under the CAMKIIa promoter, allowing investigation of TDP-43 seeding, aggregation, and propagation following inoculation with pathological TDP-43 extracts (Porta, 2018).
AAV-TDP-43 Models (AAV5, AAV8, AAV9)
AAV-mediated delivery of TDP-43 variants induces region-specific cytoplasmic aggregation or overexpression, recapitulating features such as corticospinal tract degeneration, hypothalamic atrophy, motor dysfunction, social behavior deficits, and neuromuscular pathology (Jackson, 2015; Bergh, 2025; Mori, 2025).
C9orf72 Repeat Expansion Models
Hexanucleotide (G4C2) repeat expansion in C9orf72 is the most common genetic cause of familial FTD and ALS. Disease pathology arises from toxic RNA foci formation, dipeptide repeat (DPR) protein aggregation, and haploinsufficiency, which together impair autophagy and neuroinflammatory regulation (Batra, 2017; Lopez-Herdoiza, 2023).
To learn more about the role of autophagy in neurodegeneration, see our Resource: Autophagy and Neurodegenerative Diseases.
Key C9orf72 Mouse Models
AAV(G4C2)66 and AAV(G4C2)102
Intracranial delivery of expanded G4C2 repeats induces RNA foci, DPR aggregates, cortical neuron loss, gliosis, and neuromuscular junction (NMJ) abnormalities.
Behavior: Mice show increased anxiety-like behavior, social deficits, motor coordination impairments, and memory dysfunction (Chew, 2015; Herranz-Martin, 2017).
BAC-C9-500
This model expresses 500 human G4C2 repeats using a bacterial artificial chromosome, resulting in widespread DPR aggregation, TDP-43 pathology, extensive neuronal loss, and gliosis.
Behavior: Mice develop anxiety-like behavior, motor deficits, clasping, and hindlimb paralysis (Liu, 2016).
miR- C9orf72 Knockdown
Lentiviral-mediated knockdown of C9orf72 mimics patient haploinsufficiency, leading to autophagy defects, cytoplasmic TDP-43 accumulation, synaptic loss, and late-stage neuromuscular junction abnormalities.
Behavior: Mice exhibit social interaction deficits, and depression-like behavior (Lopez-Herdoiza, 2023).
Biospective provides industry-leading staining of the neuromuscular junction and image analysis of innervation and denervation. For more information, visit: Neuromuscular Junction (NMJ) Analysis of Muscle Specimens from Animal Models.
GRN (Progranulin) Models
Progranulin is a lysosomal protein primarily produced by microglia and is essential for normal lysosomal function (Root, 2021). Mutations in GRN account for approximately 5% of FTD cases and result in progranulin haploinsufficiency or loss of function.
Key GRN Mouse Models
GRN-/- (Homozygous)
Complete loss of progranulin leads to severe lysosomal dysfunction, lipofuscinosis, microgliosis, and astrogliosis.
Behavior: Mice display compulsive grooming and reduced sociability, while hippocampus-dependent learning and memory remain preserved until late stages (Kashyap, 2023; Life, 2023).
GRN +/- (Heterozygous)
Partial progranulin deficiency results in relatively mild neuropathology but measurable synaptic and behavioral abnormalities (Filiano, 2013; Kashyap, 2023; Life, 2023).
Behavior: Mice show significant social deficits, impaired fear conditioning, hyperactivity, and increased repetitive digging behavior (Filiano, 2013; Kashyap, 2023; Life, 2023).
Humanized GRN -/-;GRNtg
This model expresses a single copy of the human GRN gene on a mouse progranulin-null background and closely resembles the heterozygous phenotype, with mild microgliosis emerging at later ages.
Behavior: Mice exhibit hyperactivity and increased repetitive digging (Life, 2023).
Comprehensive Overview of Frontotemporal Dementia (FTD) Mouse Models: Genetics, Pathology, and Behavioral Phenotypes
Tau-based Models (MAPT)
Mouse Model | Genetics | Pathology | Motor Function | Behavior |
Unilateral AAV-mediated overexpression of wild-type (2N4R) human tau in the substantia nigra pars compacta (SNc) of adult mice. | Phosphorylated tau inclusions in SNc neurons; substantial loss of SN dopaminergic neurons with corresponding striatal denervation; pronounced microgliosis and astrogliosis in the SNc; SNc, striatum, and midbrain atrophy. | Marked motor deficits (asymmetric motor dysfunction) due to unilateral injection in SNc. | SNc targeting produces a primarily motor phenotype with no significant cognitive deficits observed in the short term. | |
PS19 (P301S) tau transgenic background; stereotaxic injections of human tau PFFs into hippocampus and overlying cortex. | Extensive tau pathology with spreading from injection sites in a well-defined spatiotemporal pattern; pronounced microgliosis/astrogliosis. | No overt motor deficits. | No significant cognitive deficits. | |
rTg(TauP301L)4510 Transgenic | Human P301L tau expressed in the forebrain (hippocampus and neocortex), doxycycline regulatable. | Mice exhibit age-dependent NFT formation, gross forebrain atrophy, and severe neuronal loss. | Later stages show decreased ambulation and hindlimb clasping reflex. | Early impairments in spatial reference memory worsening with age; severe cognitive decline. |
AAV-TauP301L | Human P301L tau delivered via AAV intracranial vectors. | Tau hyperphosphorylation, aggregation, NFTs, synaptic dysfunction, neuroinflammation; no significant neuronal loss. | No overt motor deficits. | Hyperactivity, disinhibition, reduced exploration, fear conditioning and memory deficits. |
P301S Transgenic | Human P301S tau expressed in forebrain. | Synaptic pathology, filamentous tau lesions, progressive tau accumulation, neuronal loss, brain atrophy. | No overt motor deficits. | Early hyperactivity, reduced anxiety, spatial memory deficits. |
Pro-Aggregation Tau (hTau40, ΔK280) | Regulatable forebrain expression of pro-aggregation tau with ΔK280 mutation. | Tau aggregation, hyperphosphorylation, β-structure formation, strong reduction in hippocampal spine synapses; no neuronal loss. | No overt motor deficits. | Severe learning and memory deficits at later stages. |
P301S; Int10+3; S320F Knock-In | Triple-mutant knock-in expressing P301S, Int10+3, S320F. | Progressive tau accumulation, synaptic loss, severe brain atrophy, astrogliosis. | No overt motor deficits. | Increased anxiety, repetitive behaviors, impaired learning and behavioral flexibility. |
Summary of commonly used FTD mouse models based on Tau (MAPT) mutations. Key genetic alterations, characteristic neuropathological features (including tau aggregation and neurofibrillary pathology), motor function outcomes, and behavioral phenotypes are presented to support preclinical investigations of disease mechanisms and therapeutic strategies.
TDP-43TDP-43-based Models (TARDBP)
Mouse Model | Genetics | Pathology | Motor Function | Behavior |
Double transgenic (NEFH-tTA × tetO-hTDP-43-ΔNLS) enabling neuronal expression of human TDP-43 lacking a nuclear localization signal; expression is doxycycline-regulatable. | Cytoplasmic TDP-43 mislocalization with phosphorylated aggregates; widespread neurodegeneration; NMJ denervation; severe microgliosis and astrogliosis; hindlimb muscle atrophy. | Rapid onset of severe motor impairment upon induction. | Primarily motor phenotype with no notable cognitive deficits reported. Biospective’s Low Dox protocol slows disease progression, extending survival and enabling longitudinal assessments for therapeutic efficacy studies. | |
TDP-43 Q331K Transgenic | Human TARDBP Q331K under prion promoter. | Symptoms develop without classic cytoplasmic TDP-43 aggregates; protein remains nuclear. | Early-stage tremors, abnormal gait, reduced hindlimb mass, muscle function decline. | Working memory and cognitive flexibility deficits; spatial learning preserved. |
TDP-43 Q331K Knock-In | Physiologically relevant mutation in endogenous TARDBP. | Increased TDP-43 expression, altered RNA splicing, significant brain atrophy, ventricular enlargement, widespread microglial activation. | No overt motor deficits. | Learning, attention, and memory deficits. |
AAV5-TDP-43 | AAV-mediated human TDP-43 delivery to hypothalamus. | Dose-dependent hypothalamic atrophy, nuclear and cytoplasmic TDP-43 inclusions, metabolic dysregulation. | Reduced motor activity and coordination. | Reduced exploratory behavior, decreased nesting (apathy-like behavior). |
Overview of widely used FTD mouse models driven by TDP-43 (TARDBP) mutations or dysregulation. The table outlines major genetic modifications, hallmark pathological findings such as TDP-43 mislocalization and aggregation, motor impairments, and behavioral abnormalities relevant for mechanistic and translational research.
C9orf72 Repeat Expansion Models
Mouse Model | Genetics | Pathology | Motor Function | Behavior |
AAV(G4C2)66 | Human G4C2 66-repeat expansion via AAV. | DPRs, pTDP-43 aggregates, cortical neuron loss, neuroinflammation. | Reduced motor function. | Increased anxiety, sociability deficits. |
AAV(G4C2)102 | Human G4C2 102-repeat expansion via AAV. | RNA foci, DPR pathology, Purkinje cell apoptosis, sparse cytoplasmic TDP-43 aggregates. | Reduced coordination, decreased activity, worsening gait with age. | Working memory deficits. |
BAC-C9-500 | BAC transgenic expressing human C9orf72 with 500 repeats. | Widespread DPR aggregation, pTDP-43 pathology, extensive neuronal loss in cortex, hippocampus, spinal cord; neuroinflammation. | Gait abnormalities, reduced grip strength, hindlimb clasping, paralysis. | Increased anxiety. |
miR-C9orf72 | Lentiviral knockdown to mimic haploinsufficiency. | Autophagy-lysosomal dysfunction, TDP-43 aggregates, reduced cortical synaptic density. | Reduced muscular strength, neuromuscular junction abnormalities at later stages. | Early deficits in social interaction, increased depression-like behavior. |
Summary of FTD mouse models based on C9orf72 repeat expansion models. Listed are the principal genetic strategies, repeat-associated pathological features (including RNA foci formation and dipeptide repeat protein accumulation), motor phenotypes, and behavioral outcomes to facilitate preclinical therapeutic development.
Progranulin-based Models (GRN)
Mouse Model | Genetics | Pathology | Motor Function | Behavior |
Homozygous Progranulin-Null (GRN-/-) | Complete loss of progranulin. | Lysosomal dysfunction, neuroinflammation, TDP-43 aggregates. | No overt motor deficits. | Compulsive grooming, reduced sociability; hippocampal learning preserved. |
Heterozygous Progranulin-Null (GRN+/-) | One functional copy; 30–50% of wild-type progranulin. | Minimal neuropathology. | No overt motor deficits. | Social deficits, impaired fear conditioning, hyperactivity, increased repetitive behaviors. |
Humanized Progranulin-Deficient (GRN-/-; GRNtg) | Single copy of human GRN on GRN-null background. | Mild microgliosis, similar to GRN+/-. | No overt motor deficits. | Hyperactivity, increased repetitive behaviors. |
Summary of established FTD mouse models involving progranulin (GRN) deficiency. The table details genetic strategies, associated neuropathology (including lysosomal dysfunction and TDP-43 pathology), motor performance changes, and behavioral phenotypes to inform studies of disease pathogenesis and intervention approaches.
What are the advantages of AAV-Induced FTD Models?
AAV-based approaches provide a fast, flexible, and precise platform for modeling neurodegenerative diseases, such as FTD (Lunev, 2022; Aliev, 2025):
Rapid and Flexible: AAV delivery avoids breeding requirements, shortens experimental timelines, and allows combination with existing transgenic or knockout lines. High-throughput studies allow for drug screening.
Targeted Expression: AAV vectors enable region- and cell type-specific gene expression that can be temporally controlled and highly reproducible.
Experimental Precision: Gene delivery in adult animals minimizes developmental confounds and allows baseline phenotyping prior to intervention.
Safety: Recombinant AAVs are non-pathogenic, efficiently transduce non-dividing cells, and support long-term transgene expression.
In summary, AAV-induced FTD models accelerate preclinical research and provide a powerful platform for dissecting disease mechanisms and evaluating therapeutic strategies.
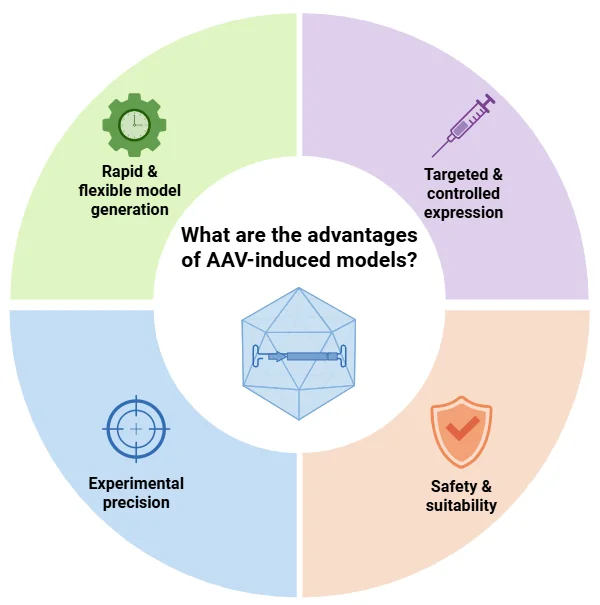
AAV-induced models enable rapid and precise generation of tailored disease models, supporting targeted gene expression, high experimental reproducibility, and a strong safety profile for translational neurodegenerative research.
What in-life imaging techniques support FTD research?
Neuroimaging plays a critical role in advancing our understanding of FTD, providing non-invasive biomarkers for early diagnosis and longitudinal insights into disease progression. Techniques including structural magnetic resonance imaging (MRI), [18F]fluorodeoxyglucose positron emission tomography ([18F]FDG PET), diffusion tensor imaging (DTI), and resting-state functional MRI (rs-fMRI) have consistently revealed subtype-specific patterns of atrophy, hypometabolism, and network disruption.
In bvFTD, structural MRI and FDG PET typically demonstrate pronounced atrophy and hypometabolism in the prefrontal cortex and anterior temporal lobes, with relative preservation of posterior brain regions (Bruun, 2019; Peet, 2021). Brain volume loss may precede clinical onset and progress at rates up to 3% annually, particularly in the frontal lobes. Frontal lobe atrophy correlates with executive dysfunction, while temporal lobe degeneration is linked to episodic memory impairment (Ghetti, 2015; Whitwell, 2019). DTI studies reveal white matter degeneration in fronto-striatal and fronto-thalamic tracts, which correlates with behavioral severity. Resting-state fMRI demonstrates reduced connectivity within the salience network, corresponding to both behavioral and executive dysfunction. As FTD advances, connectivity declines extend to broader networks, including frontal, basal ganglia, and dorsal attention systems (Ferreira, 2022).
Preclinical imaging in FTD mouse models recapitulates many features of human pathology. In rTg4510 tauopathy mice, structural MRI detects age-dependent cortical and hippocampal atrophy, while DTI identifies early white matter disorganization as early as 2.5 months, with disordered axonal architecture evident by 8 months (Sahara, 2017). The TDP-43Q331K knock-in model exhibits atrophy in the frontal, entorhinal, motor, orbital, and cingulate cortices, as well as the dentate gyrus and thalamus. These changes mirror early-stage ALS-FTD, and patterns observed in patients carrying C9orf72, MAPT, or GRN mutations. Ventricular enlargement in these mice reaches 49.7%, reflecting presymptomatic patterns observed in human genetic FTD carriers (Lin, 2021).
Complementary metabolic imaging in the TDP-43A315T model using longitudinal [18F]FDG PET reveals region-specific alterations in glucose metabolism, closely paralleling human ALS-FTD. Hypometabolism is observed in the unilateral motor and somatosensory cortices and striatum, likely reflecting synaptic and mitochondrial dysfunction or neuronal loss. In contrast, hypermetabolism is detected in the bilateral substantia nigra, reticular nucleus, and amygdaloid nucleus, potentially indicative of neuroinflammation and microglial activation. These findings align with functional biomarkers of early ALS-FTD, particularly the focal, asymmetric onset of motor dysfunction seen in approximately 98% of ALS patients (Weerasekera, 2020).
At Biospective, our fully automated image processing pipeline, PIANOTM, has been used to analyze structural and diffusion MRI in FTD research. This platform enables disease-specific intervention targeting while minimizing the number of trial participants required, maintaining statistical power. Studies demonstrate that multimodal imaging – combining structural, diffusion, and metabolic data – provides robust tools for differential diagnosis, disease monitoring, and patient stratification in clinical trials (Whitwell, 2019).
For further insights, see to our Innovation Presentations: Frontotemporal Dementia (FTD) & MRI Brain Atrophy, Diffusion MRI & Frontotemporal Dementia (FTD), and Brain Atrophy Analysis in Mouse Models of Neurodegeneration.
FAQs
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on Frontotemporal Dementia (FTD) and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
TDP-43: Role in ALS and Frontotemporal Dementia (FTD)
An overview of TDP-43, its physiological role, significance in ALS and FTD pathology, and therapeutic strategies involving TDP-43.
Neuroimaging in Frontotemporal Dementia & Clinical Trials
The utility of MRI & PET imaging biomarkers in our understanding of Frontotemporal Dementia (FTD) variants, and their use as endpoints in FTD clinical trials.
A Guide to ALS Models for Drug Discovery
A Resource for the most effective use of research animal models (mouse & rat models) of Amyotrophic Lateral Sclerosis (ALS) for preclinical testing of therapeutics.