Guide des modèles murins de démence frontotemporale (DFT)
Une ressource offrant un aperçu complet des modèles animaux utilisés dans la recherche sur la démence frontotemporale, y compris les mécanismes génétiques et pathologiques de la maladie.
Table des matières
Qu'est-ce que la démence frontotemporale (DFT) ?
Caractéristiques cliniques et sous-types
La démence frontotemporale (DFT) est un type courant de démence, qui touche particulièrement les personnes âgées de moins de 65 ans. La maladie d'Alzheimer est la cause la plus fréquente de démence précoce , suivie de la démence vasculaire et de la dégénérescence lobaire frontotemporale (DLFT) (Vieira, 2013). La DFT comprend un groupe cliniquement et pathologiquement hétérogène de syndromes neurodégénératifs associés à une dégénérescence progressive des lobes frontaux et temporaux. Sur le plan clinique, la DFT représente la manifestation symptomatique de la pathologie FTLD sous-jacente et se caractérise par des troubles progressifs du comportement, des fonctions exécutives et/ou du langage (Root, 2021).
La DFT est généralement classée en syndromes cliniques distincts en fonction des symptômes prédominants au moment de l'apparition de la maladie. La présentation la plus courante est la variante comportementale de la DFT (bvFTD), qui représente environ 60 à 80 % des cas. La variante comportementale de la DFT se caractérise par des changements précoces et progressifs de la personnalité et de la conduite sociale, notamment une désinhibition comportementale, une apathie, des comportements compulsifs ou stéréotypés et une perte de discernement. Au fur et à mesure de l'évolution de la maladie, les patients peuvent également présenter des troubles du langage, des caractéristiques parkinsoniennes ou une maladie du motoneurone (MND).
Les deux principales variantes linguistiques de la DFT sont classées sous le terme d'aphasie primaire progressive (APP) :
La variante sémantique de l'APP (svPPA) se caractérise par une perte progressive des connaissances sémantiques, y compris une altération de la compréhension des mots et une diminution de la reconnaissance des objets et des visages. Les patients présentent typiquement un discours fluide mais vide et une anomie sévère.
La variante non fluente/agrammatique de l'APP (nfvPPA) se définit par un discours non fluent et sans effort. Les caractéristiques cliniques peuvent inclure l'agrammatisme dans le langage parlé et écrit et l'apraxie de la parole - un trouble de la planification motrice qui perturbe la production de mots(Whitwell, 2019).
Malgré les progrès réalisés dans notre compréhension du spectre clinique et des fondements génétiques de la DFT, il n'existe actuellement aucun traitement de fond approuvé. Les traitements existants sont symptomatiques et visent à gérer les symptômes comportementaux ou cognitifs, plutôt qu'à modifier la progression de la maladie, ce qui souligne la nécessité de disposer de modèles précliniques robustes.
Neuropathologie et génétique
La DFT est hétérogène sur le plan neuropathologique et est le plus souvent définie par l'agrégation anormale de l'une des trois protéines suivantes : la protéine tau associée aux microtubules, la protéine TAR de liaison à l'ADN 43 (TDP-43) ou la protéine fusionnée dans le sarcome (FUS). Sur la base de la protéine pathologique prédominante, la DFT est sous-classée en FTLD-tau, FTLD-TDP et FTLD-FUS.
Il est important de noter que la présentation clinique ne permet pas de prédire de manière fiable la pathologie moléculaire sous-jacente. Par exemple, la bvFTD peut être associée à n'importe laquelle des trois principales formes pathologiques. En revanche, la svPPA est le plus souvent liée à la FTLD-TDP, tandis que la nfvPPA est principalement associée à la pathologie FTLD-tau.
Une proportion importante des cas de DFT sont familiaux et suivent généralement un modèle d'hérédité autosomique dominante. Les gènes les plus fréquemment impliqués sont MAPT, GRN (progranuline) et une expansion de répétitions hexanucléotidiques GGGGCC dans C9orf72. Alors que les mutations MAPT sont associées à la FTLD-tau, les mutations GRN et C9orf72 sont liées à la pathologie FTLD-TDP. Les trois mutations se présentent généralement sous la forme d'une DFT-b, bien que les porteurs des mutations GRN et C9orf72 présentent souvent une plus grande variabilité phénotypique, y compris des déficits du langage, le parkinsonisme ou la sclérose latérale amyotrophique (SLA) (Whitwell, 2019).
Pour en savoir plus sur les caractéristiques neuropathologiques de la DFT, consultez notre ressource : La neuro-imagerie dans la démence frontotemporale et les essais cliniques.
Il est désormais largement reconnu que la DFT et la SLA constituent un continuum de maladies neurodégénératives, partageant des caractéristiques génétiques, cliniques et pathologiques qui se chevauchent. Le lien génétique le plus important entre les deux maladies est l'expansion de la répétition C9orf72, avec des mutations moins fréquentes dans TARDBP, SQSTM1, UBQLN2, CHMP2B, CHCHD10 et VCP, ce qui confirme l'existence d'un spectre pathogénique commun (Lopez-Herdoiza, 2023; Genin, 2024).
Pour en savoir plus sur la SLA, consultez notre ressource : Guide des modèles de SLA pour la découverte de médicaments.
Pourquoi les souris sont-elles utilisées comme modèles animaux de la DFT ?
Compte tenu de l'hétérogénéité clinique et pathologique de la DFT, un large éventail de modèles de souris transgéniques et à médiation AAV ont été développés pour récapituler les principales caractéristiques moléculaires et neuropathologiques de la maladie. Les souris restent l'espèce la plus utilisée dans la recherche sur les DFT en raison de leur facilité d'adaptation génétique, de la conservation des circuits neuronaux et de leur aptitude à des investigations moléculaires, cellulaires et fonctionnelles détaillées. La plupart des lignées de souris atteintes de DFT sont conçues pour exprimer des mutations ou des protéines associées à la maladie, ce qui permet de réaliser des études mécanistiques au niveau des gènes et des protéines en rapport avec la pathogenèse de la DFT (Ahmed, 2017).
La DFT affecte principalement le réseau de saillance, qui comprend l'insula antérieure, le cortex cingulaire antérieur, le striatum ventral, l'amygdale, le thalamus dorsomédial, l'hypothalamus et plusieurs noyaux du tronc cérébral, notamment le gris périaqueducal, la substantia nigra et l'aire tegmentale ventrale. Ces régions interconnectées régissent des processus physiologiques et comportementaux fondamentaux tels que la douleur, la faim, la soif, la régulation autonome, le traitement de la récompense, la motivation et les réactions de peur. Il est important de noter que l'architecture centrale et la connectivité de ce réseau sont très conservées d'une espèce à l'autre, ce qui justifie l'utilisation de souris pour modéliser les aspects clés du dysfonctionnement des circuits liés à la DFT (Roberson, 2012).
Domaines comportementaux affectés par la DFT et leur pertinence pour les modèles murins
Plusieurs domaines comportementaux perturbés dans la DFT reposent sur des circuits neuroanatomiques conservés et peuvent donc être partiellement modélisés chez les rongeurs à l'aide de paradigmes comportementaux validés (Kesner, 2011; Bizon, 2012; Roberson, 2012; Hamilton, 2015; Mora, 2023):
Dysfonctionnement social : La réduction de l'engagement social et la perte d'empathie dans la DFT sont liées au dysfonctionnement du cortex préfrontal médian (PFC). Des déficits comparables sont observés chez les souris mutantes présentant une atteinte du CPF dorsomédian. Le cortex cingulaire antérieur - également impliqué dans la DFT - joue un rôle clé dans la modulation des comportements empathiques et sociaux chez les rongeurs.
Comportements répétitifs: Le toilettage pathologique et les comportements stéréotypés observés dans certains modèles de souris knock-out correspondent aux comportements compulsifs et répétitifs observés dans la DFT. Le striatum, impliqué dans les deux espèces, joue un rôle central dans ces comportements.
Dysrégulation émotionnelle : Les patients atteints de DFT présentent souvent des réactions émoussées aux émotions négatives, associées à l'atrophie de l'amygdale. De même, les souris dont le fonctionnement de l'amygdale est perturbé présentent des troubles du conditionnement de la peur, ce qui confirme la valeur translationnelle de ces modèles.
Déficits exécutifs : Les troubles progressifs de la planification, de la prise de décision et de la mémoire de travail sont des caractéristiques essentielles de la DFT et impliquent un dysfonctionnement du PFC dorsolatéral, du cortex cingulaire antérieur, du PFC médian et des ganglions de la base. Dans les modèles murins de DFT, la perturbation du circuit PFC- striatal-thalamique entraîne également des dysfonctionnements exécutifs. Ces déficits sont généralement évalués à l'aide de tâches telles que le labyrinthe en T, le labyrinthe en Y et les paradigmes de déplacement de l'attention.
Flexibilité cognitive: Les difficultés à modifier l'attention ou le comportement et la rigidité de la pensée dans la DFT sont liées au cortex orbitofrontal (OFC), au PFC, au thalamus médiodorsal et aux ganglions de la base. L'atrophie du cortex orbitofrontal est associée à la rigidité comportementale chez les patients atteints de DFT et chez les rongeurs. Les tests d'apprentissage inversé et de déplacement opérant sur écran tactile sont utilisés pour mesurer l'inflexibilité cognitive.
Dysfonctionnement moteur : Les symptômes moteurs se manifestent surtout dans les cas de DFT-SAL ou de DFT avec mutations tau impliquant le cortex moteur (aire motrice primaire et supplémentaire), les ganglions de la base et les voies motrices du tronc cérébral. Dans les modèles de rongeurs atteints de DFT, les phénotypes moteurs sont liés à la dégénérescence du cortex et des ganglions de la base. Le rotarod, la force de préhension, l'analyse de la marche et la préhension sont couramment utilisés pour quantifier le déclin moteur.
Pour en savoir plus sur les tests comportementaux et moteurs couramment utilisés pour suivre la progression de la maladie, voir : Tests des fonctions motrices et sensorielles et Tests du sommeil et de la cognition.
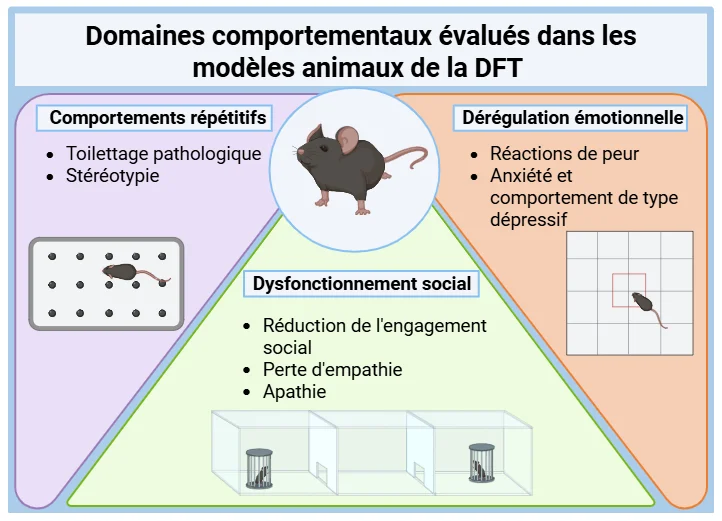
Les évaluations comportementales complètes des modèles murins de DFT, comprenant des mesures des comportements répétitifs, des interactions sociales et de la réactivité émotionnelle, fournissent des critères d'évaluation solides pour l'évaluation préclinique des traitements modificateurs de la maladie.
Considérations importantes concernant les modèles de souris dans la recherche sur les DFT
Les modèles murins de DFT ont été principalement conçus pour reproduire les principales caractéristiques comportementales de la DFTB, notamment le retrait social, la désinhibition, les comportements compulsifs et les schémas moteurs stéréotypés. Cet objectif est à la fois pratique et cliniquement pertinent, car la bvFTD est le sous-type de DFT le plus répandu, survenant environ quatre fois plus fréquemment que les APP (Hogan, 2016).
Ces modèles précliniques se sont révélés inestimables pour découvrir les mécanismes de la maladie, cartographier les circuits neuronaux affectés et identifier des cibles thérapeutiques potentielles dans des systèmes accessibles et bien caractérisés. Cependant, la modélisation des formes de DFT à prédominance linguistique - svPPA et nfvPPA - reste un défi majeur. Ces syndromes reposent sur des réseaux linguistiques et sémantiques complexes qui n'ont pas d'homologues directs dans le cerveau des rongeurs, ce qui rend impossible la récapitulation complète des déficits liés au langage chez la souris.
Par conséquent, la recherche préclinique a mis l'accent de manière appropriée sur les dimensions comportementales qui sont à la fois fortement altérées dans la DFT et expérimentalement réalisables dans les modèles animaux. Pour pallier les limites des systèmes de rongeurs, les chercheurs intègrent de plus en plus des approches humaines complémentaires, telles que les neurones dérivés de cellules souches pluripotentes induites (iPSC), les organoïdes cérébraux et les études de neuro-imagerie des patients, afin d'étudier les aspects du langage et de la pathologie du réseau sémantique (Roberson, 2012; Whitwell, 2019).
Malgré ces contraintes, les modèles de souris restent une composante essentielle du pipeline de la recherche translationnelle. Ils fournissent des informations essentielles sur les mécanismes cellulaires et circulaires précoces de la DFT et offrent une plateforme robuste pour évaluer les traitements candidats in vivo.
Quels sont les principaux modèles de souris DFT utilisés en recherche préclinique ?
Modèles basés sur la protéine tau (MAPT)
Les mutations du gène tau(MAPT) sont l'une des principales causes génétiques de la DFT, au même titre que les mutations du GRN et de C9orf72. Plus de 40 mutations MAPT associées à la DFT ont été identifiées, la plupart étant des mutations faux-sens dans la région de liaison aux microtubules. Ces mutations produisent à la fois des effets de perte de fonction, en réduisant la stabilisation des microtubules, et des effets de gain de fonction, en augmentant l'agrégation et l'hyperphosphorylation de la protéine tau (Roberson, 2012).
Les formes mutantes de la protéine tau humaine, notamment P301S et P301L, sont associées à des tauopathies telles que la FTD avec parkinsonisme-17 (FTDP-17).
Chez Biospective, nous générons des modèles de souris Tau en utilisant plusieurs approches :
Ensemencement de fibrilles préformées de Tau (PFF): Permet la propagation de tau pathologique in vivo, facilitant les études sur la propagation et l'agrégation de tau. Explorez le modèle ici : Modèles de propagation des fibrilles de tau.
Diffusion d'AAV-hTau : L'injection intracrânienne de vecteurs AAV dans le cerveau de souris C57BL/6 adultes récapitule les principales caractéristiques de la tauopathie humaine, notamment les agrégats de tau phosphorylés, le dysfonctionnement comportemental et l'atrophie cérébrale détectable par IRM in vivo. Cette approche permet de générer des résultats dans un délai relativement court. Voir notre présentation d'images interactives ici : Modèles de souris AAV Tau.
Principaux modèles de souris Tau
rTg(TauP301L)4510
Ce modèle exprime la mutation tau humaine P301L dans le cerveau antérieur, y compris l'hippocampe et le néocortex, avec une expression du transgène qui peut être régulée par la doxycycline. Les souris présentent une formation d'enchevêtrements neurofibrillaires (ENF) dépendante de l'âge, accompagnée d'une atrophie prononcée du cerveau antérieur et d'une perte de neurones.
Comportement : Les souris présentent une hyperactivité précoce et un comportement anxieux réduit, suivis de déficits de la mémoire spatiale à partir de l'âge de quatre mois et de déficiences motrices progressives, y compris un réflexe d'agrippement caractéristique (Lewis, 2000; Ramsden, 2005; Pennanen, 2006).
AAV-TauP301L
Dans ce modèle, la protéine tau humaine P301L est administrée par voie intracrânienne à l'aide de vecteurs AAV, ce qui entraîne une expression généralisée de la protéine tau dans les régions cérébrales ciblées. Cette approche entraîne une hyperphosphorylation de la protéine tau, la formation de pré-tangles et de NFT matures, de neurites dystrophiques et de réponses neuroinflammatoires.
Comportement : Les souris présentent une hyperactivité, une désinhibition, une altération du conditionnement de la peur et des déficits de mémoire, reflétant un dysfonctionnement des circuits dépendant de l'hippocampe et de l'amygdale (Cook, 2015; Silva-Llanes, 2025).
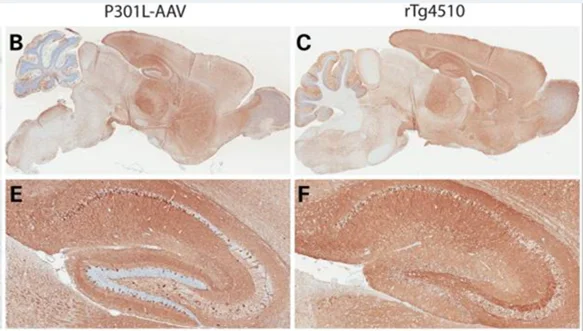.webp)
L'expression généralisée de tau humaine dans le modèle de souris AAV-TauP301L (panneaux B, E) reflète étroitement la distribution observée chez les souris rTg4510 (panneaux C, F). Figure reproduite de Cook et al. (Cook, 2015) sous la licence Creative Commons Attribution.
Tau P301S
Ce modèle transgénique(par exemple, les souris PS19) exprime la mutation tau humaine P301S et se caractérise par des troubles comportementaux et cognitifs précoces qui apparaissent avant l'apparition d'une pathologie NFT étendue. Un dysfonctionnement synaptique est observé dès l'âge de trois mois, suivi d'une accumulation progressive de tau, d'une perte neuronale et d'une atrophie cérébrale entre neuf et douze mois.
Comportement : Les phénotypes précoces comprennent l'hyperactivité et un comportement anxieux réduit, avec des déficits de mémoire spatiale détectés dans le labyrinthe aquatique de Morris (Takeuchi, 2011).
Tau pro-agrégation (hTau40, ΔK280)
Ce modèle utilise l'expression régulable dans le cerveau antérieur d'une variante pro-agrégation de la protéine tau humaine pleine longueur contenant la mutation ΔK280, qui renforce la formation de la structure β et l'agrégation de la protéine tau (Eckermann, 2007). Bien qu'aucune perte neuronale manifeste ne soit observée, les souris présentent des déficits synaptiques marqués et une potentialisation à long terme réduite.
Comportement : Après une expression prolongée du gène, les souris développent de graves troubles de l'apprentissage et de la mémoire, alors que la fonction motrice reste intacte (Van der Jeugd, 2012).
Knock-in MAPT P301S;Int10+3;S320F
Ce modèle knock-in triple mutant présente une pathologie tau robuste et précoce, une accumulation progressive de tau pathologique, une perte synaptique et une atrophie sévère de l'hippocampe, de l'hypothalamus et de l'amygdale, accompagnée d'une astrogliose.
Comportement : Les souris présentent une anxiété accrue, des comportements répétitifs et stéréotypés, une altération de l'attention soutenue, un comportement de type apathique et des déficits d'apprentissage et de flexibilité comportementale (Morito, 2025).
Modèles basés sur le TDP-43
Les inclusions TDP-43 positives représentent le substrat pathologique le plus courant dans les DFT (FTD-TDP). La TDP-43 est une protéine nucléaire de liaison à l'ARN et à l'ADN impliquée dans le traitement et la régulation de l'ARN. Les mutations pathogènes de TARDPB entraînent une mauvaise localisation de TDP-43 du noyau vers le cytoplasme, ce qui se traduit par une combinaison de perte de fonction nucléaire et de gain de fonction cytoplasmique toxique (Roberson, 2012).
En savoir plus sur le modèle de souris TDP-43ΔNLS (rNLS8) de Biospective.
Principaux modèles de souris TDP-43
TDP-43 Q331K transgénique
Ce modèle exprime la mutation humaine TARDBP Q331K sous le promoteur du prion murin, ce qui entraîne des niveaux d'expression quasi physiologiques et une régulation à la baisse de la TDP-43 endogène de la souris. La protéine reste principalement nucléaire et ne forme pas d'agrégats cytoplasmiques classiques (Wong, 2020; Watkins, 2021).
Comportement : Les souris développent des tremblements, une démarche anormale, une masse musculaire réduite et des déficits cognitifs médiés par le cortex frontal, tandis que la mémoire dépendante de l'hippocampe reste largement préservée (Wong, 2020; Watkins, 2021).
TDP-43 Q331K Knock-In
Dans ce modèle physiologiquement pertinent, la mutation Q331K est introduite dans le gène TARDBP endogène de la souris, évitant ainsi les artefacts de surexpression. Le modèle récapitule les principales modifications structurelles du cerveau observées dans la SLA-FTD, notamment l'atrophie des cortex frontal et entorhinal et de l'hippocampe, ainsi qu'une activation microgliale généralisée (White, 2018; Lin, 2021).
Comportement : Les souris présentent des troubles de l'apprentissage, de l'attention et de la mémoire (White, 2018; Lin, 2021).
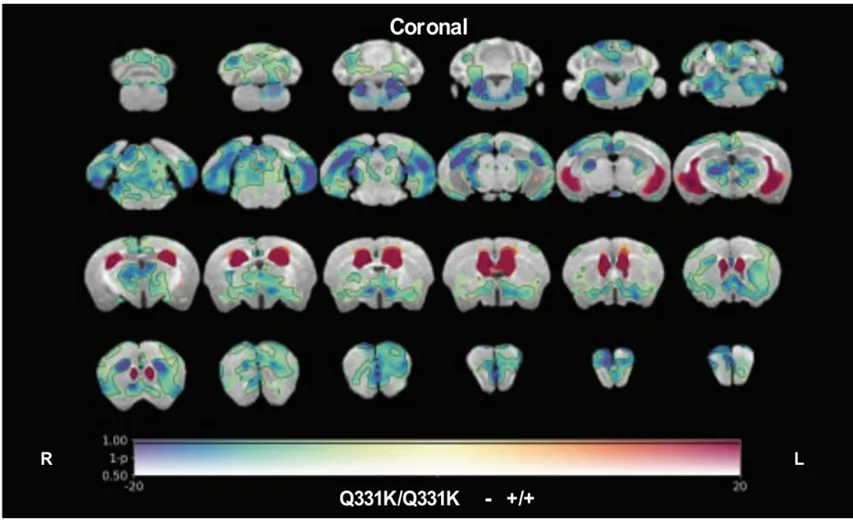.webp)
L'imagerie par résonance magnétique (IRM)in vivo révèle une perte de volume cérébral régional (couleurs froides) et un élargissement ventriculaire (couleurs chaudes) chez les souris knock-in TDP-43Q331K/Q331K par rapport aux témoins de type sauvage appariés selon l'âge, mettant en évidence des changements structurels qui reflètent la pathologie humaine de la SLA-FTD. Figure reproduite de Lin et al. (Lin, 2021) sous la licence Creative Commons Attribution.
Inoculation de CamKIIa-hTDP-43NLSm
Ce modèle utilise des souris transgéniques exprimant une forme de TDP-43 humaine mal localisée dans le cytoplasme sous le promoteur CAMKIIa, ce qui permet d'étudier l'ensemencement, l'agrégation et la propagation de TDP-43 après inoculation avec des extraits de TDP-43 pathologiques (Porta, 2018).
Modèles AAV-TDP-43 (AAV5, AAV8, AAV9)
L'administration par AAV de variantes de TDP-43 induit une agrégation cytoplasmique ou une surexpression spécifique à une région, récapitulant des caractéristiques telles que la dégénérescence du tractus corticospinal, l'atrophie hypothalamique, le dysfonctionnement moteur, les déficits du comportement social et la pathologie neuromusculaire (Jackson, 2015; Bergh, 2025; Mori, 2025).
Modèles d'expansion de la répétition C9orf72
L'expansion de répétitions hexanucléotidiques (G4C2) dans le gène C9orf72 est la cause génétique la plus fréquente des DFT familiales et de la SLA. La pathologie de la maladie résulte de la formation de foyers d'ARN toxiques, de l'agrégation de protéines à répétition dipeptidique (DPR) et de l'haploinsuffisance, qui, ensemble, altèrent l'autophagie et la régulation neuroinflammatoire (Batra, 2017; Lopez-Herdoiza, 2023).
Pour en savoir plus sur le rôle de l'autophagie dans la neurodégénérescence, consultez notre ressource :Autophagie et maladies neurodégénératives.
Principaux modèles de souris C9orf72
AAV(G4C2)66 et AAV(G4C2)102
L'administration intracrânienne de répétitions G4C2 étendues induit des foyers d'ARN, des agrégats de DPR, une perte de neurones corticaux, une gliose et des anomalies de la jonction neuromusculaire (NMJ) .
Comportement : Les souris présentent un comportement anxieux accru, des déficits sociaux, des troubles de la coordination motrice et un dysfonctionnement de la mémoire (Chew, 2015; Herranz-Martin, 2017).
BAC-C9-500
Ce modèle exprime 500 répétitions G4C2 humaines à l'aide d'un chromosome artificiel bactérien, ce qui entraîne une agrégation généralisée de la RPD, une pathologie TDP-43, une perte neuronale étendue et une gliose.
Comportement : Les souris développent un comportement de type anxieux, des déficits moteurs, une agrippement et une paralysie des membres postérieurs (Liu, 2016).
abattage du miR- C9orf72
Le knockdown de C9orf72 par voie lentivirale imite l'haploinsuffisance du patient, entraînant des défauts d'autophagie, une accumulation cytoplasmique de TDP-43, une perte synaptique et des anomalies de la jonction neuromusculaire à un stade avancé.
Comportement : Les souris présentent des déficits d'interaction sociale et un comportement de type dépressif (Lopez-Herdoiza, 2023).
Biospective fournit une coloration de la jonction neuromusculaire et une analyse d'image de l'innervation et de la dénervation qui sont à la pointe de l'industrie. Pour plus d'informations, visitez le site : Analyse de la jonction neuromusculaire (NMJ) de spécimens musculaires provenant de modèles animaux.
Modèles GRN (Progranuline)
La progranuline est une protéine lysosomale principalement produite par la microglie et essentielle à la fonction lysosomale normale (Root, 2021). Les mutations de la GRN sont à l'origine d'environ 5 % des cas de DFT et entraînent une haploinsuffisance ou une perte de fonction de la progranuline.
Principaux modèles de souris GRN
GRN-/- (homozygote)
La perte totale de la progranuline entraîne un dysfonctionnement lysosomal grave, une lipofuscinose, une microgliose et une astrogliose.
Comportement : Les souris présentent un toilettage compulsif et une sociabilité réduite, tandis que l'apprentissage et la mémoire dépendant de l'hippocampe restent préservés jusqu'à un stade avancé (Kashyap, 2023; Life, 2023).
GRN +/- (Hétérozygote)
La déficience partielle en progranuline entraîne une neuropathologie relativement légère, mais des anomalies synaptiques et comportementales mesurables (Filiano, 2013; Kashyap, 2023; Life, 2023).
Comportement : Les souris présentent des déficits sociaux significatifs, un conditionnement de la peur altéré, une hyperactivité et un comportement de fouille répétitif accru (Filiano, 2013; Kashyap, 2023; Life, 2023).
GRN humanisé -/-;GRNtg
Ce modèle exprime une seule copie du gène GRN humain sur un fond de souris progranuline-nulle et ressemble étroitement au phénotype hétérozygote, avec une légère microgliose apparaissant à un âge plus avancé.
Comportement : Les souris présentent une hyperactivité et un nombre accru de fouilles répétitives (Life, 2023).
Vue d'ensemble des modèles de souris de la démence frontotemporale (DFT) : Génétique, pathologie et phénotypes comportementaux
Modèles basés sur la tau (MAPT)
Modèle de souris | Génétique | Pathologie | Fonction motrice | Comportement |
Surexpression unilatérale par AAV de la protéine tau humaine de type sauvage (2N4R) dans la substantia nigra pars compacta (SNc) de souris adultes. | Inclusions de tau phosphorylée dans les neurones de la SNc ; perte substantielle de neurones dopaminergiques de la SN avec dénervation correspondante du striatum ; microgliose et astrogliose prononcées dans la SNc ; atrophie de la SNc, du striatum et du mésencéphale. | Déficits moteurs marqués (dysfonctionnement moteur asymétrique) dus à l'injection unilatérale dans le SNc. | Le ciblage du SNc produit un phénotype principalement moteur, sans déficits cognitifs significatifs observés à court terme. | |
Fond transgénique tau PS19 (P301S) ; injections stéréotaxiques de PFFs tau humaines dans l'hippocampe et le cortex sus-jacent. | Pathologie tau étendue se propageant à partir des sites d'injection selon un schéma spatio-temporel bien défini ; microgliose/astrogliose prononcée. | Pas de déficits moteurs manifestes. | Pas de déficits cognitifs significatifs. | |
rTg(TauP301L)4510 Transgénique | Tau humaine P301L exprimée dans le cerveau antérieur (hippocampe et néocortex), régulée par la doxycycline. | Les souris présentent une formation de NFT en fonction de l'âge, une atrophie du cerveau antérieur et une perte neuronale sévère. | Les stades ultérieurs montrent une diminution de l'ambulation et du réflexe d'agrippement des membres postérieurs. | Les troubles précoces de la mémoire de référence spatiale s'aggravent avec l'âge ; déclin cognitif sévère. |
AAV-TauP301L | Tau humaine P301L délivrée par des vecteurs intracrâniens AAV. | Hyperphosphorylation de la protéine Tau, agrégation, NFT, dysfonctionnement synaptique, neuroinflammation ; pas de perte neuronale significative. | Pas de déficits moteurs manifestes. | Hyperactivité, désinhibition, réduction de l'exploration, conditionnement à la peur et déficits de mémoire. |
P301S transgénique | Tau humaine P301S exprimée dans le cerveau antérieur. | Pathologie synaptique, lésions tau filamenteuses, accumulation progressive de tau, perte neuronale, atrophie cérébrale. | Pas de déficits moteurs manifestes. | Hyperactivité précoce, anxiété réduite, déficits de la mémoire spatiale. |
Tau pro-agrégation (hTau40, ΔK280) | Expression régulable dans le cerveau antérieur de la protéine tau pro-agrégation avec la mutation ΔK280. | Agrégation de la protéine Tau, hyperphosphorylation, formation d'une structure β, forte réduction des synapses de la colonne vertébrale de l'hippocampe ; pas de perte neuronale. | Pas de déficits moteurs manifestes. | Graves déficits d'apprentissage et de mémoire aux stades ultérieurs. |
Knock-In P301S ; Int10+3 ; S320F | Inclusion d'un triple mutant exprimant P301S, Int10+3, S320F. | Accumulation progressive de tau, perte synaptique, atrophie cérébrale sévère, astrogliose. | Pas de déficits moteurs manifestes. | Augmentation de l'anxiété, comportements répétitifs, altération de l'apprentissage et de la flexibilité comportementale. |
Résumé des modèles murins de DFT couramment utilisés, basés sur des mutations de la protéine Tau (MAPT). Les principales altérations génétiques, les caractéristiques neuropathologiques (y compris l'agrégation de tau et la pathologie neurofibrillaire), les résultats de la fonction motrice et les phénotypes comportementaux sont présentés pour soutenir les recherches précliniques sur les mécanismes de la maladie et les stratégies thérapeutiques.
Modèles basés sur la TDP-43TDP-43 (TARDBP)
Modèle de souris | Génétique | Pathologie | Fonction motrice | Comportement |
Double transgénique (NEFH-tTA × tetO-hTDP-43-ΔNLS) permettant l'expression neuronale de la TDP-43 humaine dépourvue de signal de localisation nucléaire ; l'expression est régulée par la doxycycline. | Mauvaise localisation cytoplasmique de la TDP-43 avec des agrégats phosphorylés ; neurodégénérescence généralisée ; dénervation de la NMJ ; microgliose et astrogliose sévères ; atrophie musculaire des membres postérieurs. | Apparition rapide d'une déficience motrice sévère dès l'induction. | Phénotype essentiellement moteur, aucun déficit cognitif notable n'ayant été signalé. Le protocole Low Dox de Biospective ralentit la progression de la maladie, prolongeant la survie et permettant des évaluations longitudinales pour les études d'efficacité thérapeutique. | |
TDP-43 Q331K Transgénique | TARDBP humain Q331K sous le promoteur du prion. | Les symptômes se développent sans les agrégats cytoplasmiques classiques de TDP-43 ; la protéine reste nucléaire. | Tremblements précoces, démarche anormale, réduction de la masse des membres postérieurs, déclin de la fonction musculaire. | Déficits de la mémoire de travail et de la flexibilité cognitive ; l'apprentissage spatial est préservé. |
TDP-43 Q331K Knock-In | Mutation physiologiquement pertinente de la TARDBP endogène. | Augmentation de l'expression de la TDP-43, modification de l'épissage de l'ARN, atrophie cérébrale significative, hypertrophie ventriculaire, activation microgliale généralisée. | Pas de déficits moteurs manifestes. | Déficits d'apprentissage, d'attention et de mémoire. |
AAV5-TDP-43 | Transfert de TDP-43 humain dans l'hypothalamus par l'intermédiaire d'AAV. | Atrophie hypothalamique dose-dépendante, inclusions nucléaires et cytoplasmiques de TDP-43, dérèglement métabolique. | Réduction de l'activité motrice et de la coordination. | Diminution du comportement exploratoire, diminution de la nidification (comportement de type apathique). |
Vue d'ensemble des modèles murins de DFT largement utilisés, basés sur des mutations ou une dysrégulation de la protéine TDP-43 (TARDBP). Le tableau présente les principales modifications génétiques, les résultats pathologiques caractéristiques tels que la mauvaise localisation et l'agrégation de TDP-43, les déficiences motrices et les anomalies comportementales pertinentes pour la recherche mécaniste et translationnelle.
Modèles d'expansion de la répétition C9orf72
Modèle de souris | Génétique | Pathologie | Fonction motrice | Comportement |
AAV(G4C2)66 | Expansion des 66 répétitions du gène G4C2 humain par l'intermédiaire d'AAV. | RPD, agrégats de pTDP-43, perte de neurones corticaux, neuroinflammation. | Fonction motrice réduite. | Augmentation de l'anxiété, déficits de sociabilité. |
AAV(G4C2)102 | Expansion de la réplique 102 du gène G4C2 humain par l'intermédiaire d'AAV. | Foyers d'ARN, pathologie DPR, apoptose des cellules de Purkinje, agrégats cytoplasmiques épars de TDP-43. | Coordination réduite, activité réduite, aggravation de la démarche avec l'âge. | Déficits de la mémoire de travail. |
BAC-C9-500 | BAC transgénique exprimant la protéine humaine C9orf72 avec 500 répétitions. | Agrégation généralisée de DPR, pathologie pTDP-43, perte neuronale importante dans le cortex, l'hippocampe et la moelle épinière ; neuroinflammation. | Anomalies de la démarche, réduction de la force de préhension, agrippement des membres postérieurs, paralysie. | Augmentation de l'anxiété. |
miR-C9orf72 | Knockdown lentiviral pour imiter l'haploinsuffisance. | Dysfonctionnement de l'autophagie et des lysosomes, agrégats de TDP-43, densité synaptique corticale réduite. | Réduction de la force musculaire, anomalies de la jonction neuromusculaire à des stades plus avancés. | Déficits précoces dans les interactions sociales, augmentation des comportements de type dépressif. |
Résumé des modèles murins de DFT basés sur les modèles d'expansion de la répétition de C9orf72. Les principales stratégies génétiques, les caractéristiques pathologiques associées aux répétitions (y compris la formation de foyers d'ARN et l'accumulation de protéines dipeptidiques répétées), les phénotypes moteurs et les résultats comportementaux sont énumérés afin de faciliter le développement thérapeutique préclinique.
Modèles basés sur la progranuline (GRN)
Modèle de souris | Génétique | Pathologie | Fonction motrice | Comportement |
Progranuline homozygote nulle (GRN-/-) | Perte totale de progranuline. | Dysfonctionnement lysosomal, neuroinflammation, agrégats TDP-43. | Pas de déficits moteurs manifestes. | Toilettage compulsif, sociabilité réduite ; apprentissage hippocampique préservé. |
Progranuline hétérozygote nulle (GRN+/-) | Une copie fonctionnelle ; 30 à 50 % de la progranuline de type sauvage. | Neuropathologie minimale. | Pas de déficits moteurs manifestes. | Déficits sociaux, altération du conditionnement de la peur, hyperactivité, augmentation des comportements répétitifs. |
Progranuline humanisée déficiente (GRN-/- ; GRNtg) | Copie unique de la GRN humaine sur fond de GRN-null. | Légère microgliose, similaire à GRN+/-. | Pas de déficits moteurs manifestes. | Hyperactivité, augmentation des comportements répétitifs. |
Résumé des modèles murins de DFT établis impliquant une déficience en progranuline (GRN). Le tableau détaille les stratégies génétiques, la neuropathologie associée (y compris le dysfonctionnement lysosomal et la pathologie TDP-43), les changements de performance motrice et les phénotypes comportementaux pour informer les études sur la pathogenèse de la maladie et les approches d'intervention.
Quels sont les avantages des modèles de DFT induits par AAV ?
Les approches basées sur les AAV constituent une plateforme rapide, flexible et précise pour la modélisation des maladies neurodégénératives, telles que la DFT (Lunev, 2022; Aliev, 2025):
Rapide et flexible : L'administration d'AAV évite les exigences d'élevage, raccourcit les délais expérimentaux et permet la combinaison avec des lignées transgéniques ou knock-out existantes. Les études à haut débit permettent le criblage de médicaments.
Expression ciblée: Les vecteurs AAV permettent l'expression de gènes spécifiques à une région ou à un type de cellule, qui peut être contrôlée dans le temps et hautement reproductible.
Précision expérimentale: L'administration de gènes à des animaux adultes minimise les problèmes de développement et permet un phénotypage de base avant l'intervention.
Sécurité : Les AAV recombinants ne sont pas pathogènes, transduisent efficacement les cellules qui ne se divisent pas et permettent une expression à long terme du transgène.
En résumé, les modèles de DFT induits par les AAV accélèrent la recherche préclinique et fournissent une plate-forme puissante pour disséquer les mécanismes de la maladie et évaluer les stratégies thérapeutiques.
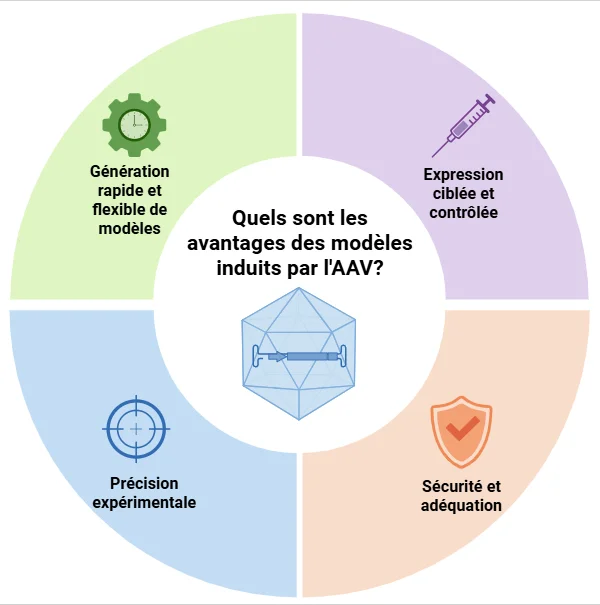
Les modèles induits par l'AAV permettent de générer rapidement et précisément des modèles de maladies sur mesure, en favorisant l'expression génétique ciblée, une reproductibilité expérimentale élevée et un profil de sécurité solide pour la recherche translationnelle sur les maladies neurodégénératives.
Quelles sont les techniques d'imagerie in vivo qui soutiennent la recherche sur les DFT ?
La neuro-imagerie joue un rôle essentiel dans l'amélioration de notre compréhension de la DFT, en fournissant des biomarqueurs non invasifs pour le diagnostic précoce et des informations longitudinales sur l'évolution de la maladie. Des techniques telles que l'imagerie par résonance magnétique (IRM) structurelle, la tomographie par émission de positons au [18F]fluorodésoxyglucose (TEP au [18F]FDG) sur le site , l'imagerie du tenseur de diffusion (DTI) et l'IRM fonctionnelle en état de repos (IRM-RS) ont systématiquement révélé des schémas d'atrophie, d'hypométabolisme et de perturbation du réseau spécifiques à un sous-type.
Dans la bvFTD, l'IRM structurelle et la TEP au FDG montrent généralement une atrophie et un hypométabolisme prononcés dans le cortex préfrontal et les lobes temporaux antérieurs, avec une préservation relative des régions cérébrales postérieures (Bruun, 2019; Peet, 2021). La perte de volume cérébral peut précéder l'apparition des symptômes et progresser à des taux allant jusqu'à 3 % par an, en particulier dans les lobes frontaux. L'atrophie du lobe frontal est corrélée à un dysfonctionnement exécutif, tandis que la dégénérescence du lobe temporal est liée à des troubles de la mémoire épisodique (Ghetti, 2015; Whitwell, 2019). Les études DTI révèlent une dégénérescence de la matière blanche dans les voies fronto-striatales et fronto-thalamiques, qui est en corrélation avec la gravité du comportement. L'IRMf à l'état de repos montre une réduction de la connectivité au sein du réseau de saillance, ce qui correspond à un dysfonctionnement comportemental et exécutif. Au fur et à mesure que la DFT progresse, les baisses de connectivité s'étendent à des réseaux plus larges, y compris les systèmes d'attention frontale, des ganglions de la base et dorsale (Ferreira, 2022).
L'imagerie préclinique des modèles murins de DFT récapitule de nombreuses caractéristiques de la pathologie humaine. Chez les souris tauopathes rTg4510, l'IRM structurelle détecte une atrophie corticale et hippocampique dépendante de l'âge, tandis que l'ITD identifie une désorganisation précoce de la substance blanche dès 2,5 mois, avec une architecture axonale désordonnée évidente à 8 mois (Sahara, 2017). Le modèle TDP-43Q331K knock-in présente une atrophie des cortex frontal, entorhinal, moteur, orbital et cingulaire, ainsi que du gyrus denté et du thalamus. Ces changements reflètent les stades précoces de la SLA-FTD et les schémas observés chez les patients porteurs de mutations C9orf72, MAPT ou GRN. L'hypertrophie ventriculaire chez ces souris atteint 49,7 %, ce qui reflète les schémas présymptomatiques observés chez les porteurs de la DFT génétique humaine (Lin, 2021).
L'imagerie métabolique complémentaire dans le modèle TDP-43A315T à l'aide de la TEP [18F]FDG longitudinale révèle des altérations du métabolisme du glucose spécifiques à certaines régions, qui sont étroitement liées à la SLA-FTD chez l'homme. Un hypométabolisme est observé dans les cortex moteur et somatosensoriel unilatéraux et dans le striatum, reflétant probablement un dysfonctionnement synaptique et mitochondrial ou une perte neuronale. En revanche, un hypermétabolisme est détecté dans la substantia nigra bilatérale, le noyau réticulaire et le noyau amygdaloïde, ce qui pourrait indiquer une neuroinflammation et une activation microgliale. Ces résultats s'alignent sur les biomarqueurs fonctionnels de la SLA-FTD précoce, en particulier l'apparition focale et asymétrique du dysfonctionnement moteur observée chez environ 98 % des patients atteints de SLA (Weerasekera, 2020).
Chez Biospective, notre pipeline de traitement d'images entièrement automatisé, PIANOTM, a été utilisé pour analyser l'IRM structurelle et de diffusion dans la recherche sur la DFT. Cette plateforme permet de cibler les interventions spécifiques à la maladie tout en minimisant le nombre de participants aux essais, ce qui maintient la puissance statistique. Les études démontrent que l'imagerie multimodale - combinant des données structurelles, de diffusion et métaboliques - fournit des outils robustes pour le diagnostic différentiel, le suivi de la maladie et la stratification des patients dans les essais cliniques (Whitwell, 2019).
Pour en savoir plus, consultez nos présentations sur l'innovation : Démence frontotemporale (DFT) et atrophie cérébrale par IRM,IRM de diffusion et démence frontotemporale (DFT), et Analyse de l'atrophie cérébrale dans les modèles murins de neurodégénérescence.
FAQ
En savoir plus sur nos modèles de maladies neurodégénératives
Contenu connexe
Informations actualisées sur la démence frontotemporale (DFT) et les meilleures pratiques relatives à l'évaluation des agents thérapeutiques dans les modèles animaux de maladies neurodégénératives.
TDP-43 - Son rôle dans la SLA et la DFT
Présentation générale de la TDP-43, de son rôle physiologique, de son importance dans la pathologie de la SLA et de la DFT, ainsi que des stratégies thérapeutiques impliquant la TDP-43.
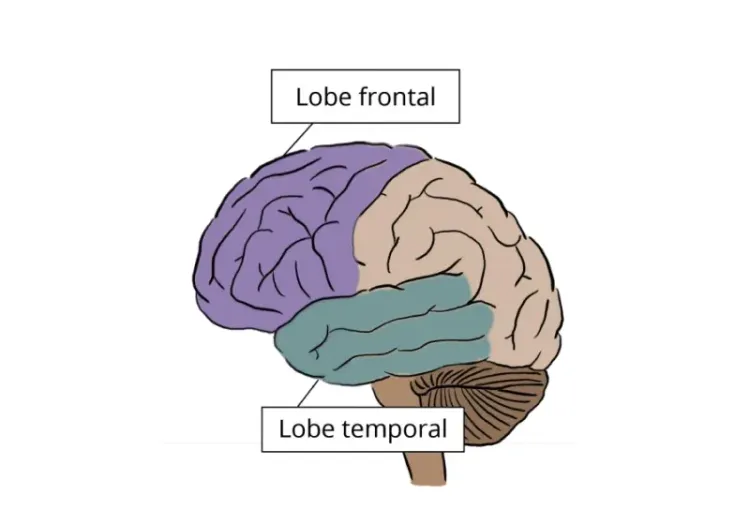
La neuroimagerie dans la démence frontotemporale et les essais cliniques
L'utilité des biomarqueurs d'imagerie IRM et TEP dans notre compréhension des variantes de la démence frontotemporale (DFT) et leur utilisation comme critères d'évaluation dans les essais cliniques sur la DFT.
Modèles de souris SLA pour le développement de médicaments
Un guide pour l'utilisation la plus efficace possible des modèles animaux de recherche sur la sclérose latérale amyotrophique (SLA) pour les essais précliniques de produits thérapeutiques.