Lysosome Dysfunction in Microglia & Astrocytes
An overview of lysosomal dysfunction in microglia & astrocytes, and its role in neurodegenerative diseases.
What is the role of lysosomes in astrocytes and microglia?
The lysosome is a cytoplasmic organelle that is primarily recognized for its role in degradation of various cellular debris (Perera, 2016). Lysosome biogenesis is primarily regulated by Transcription Factor EB (TFEB), which controls the expression of genes responsible for lysosomal proteins and enzymes. The lysosome maintains an acidic lumen via the vacuolar-ATPase (v-ATPase) proton pump, which is contained by a specialized membrane consisting of lysosomal-associated membrane proteins (LAMP). Housed in the lysosome are numerous hydrolytic enzymes that carry out its catabolic functions.
Lysosomes are important for the degradation of both extracellular and intracellular materials (Perera, 2016). Intake of extracellular materials is achieved via endocytosis, in which waste is packaged into an endosome that later fuses with the lysosome for degradation. In contrast, intracellular materials are degraded and recycled through autophagy, in which old or damaged organelles are packaged into autophagosomes that fuse with the lysosome for recycling or degradation. As such, lysosomes serve as a common terminal point in which macromolecules are broken down to their constituents for clearance or recycling.
Astrocytes and microglia are critical for supporting and regulating neuronal signaling. Astrocytes are key in recycling excess neurotransmitters to regulate neuronal signaling, as well as maintenance of the blood-brain barrier, among other functions. Microglia serve as the principal immune cells of the central nervous system (CNS). Microglia are phagocytic cells that are essential for clearance of pathogens and cellular debris, including protein aggregates found in the neurodegenerative diseases (Kreher, 2021). As the primary endpoint for degradation of cellular debris, lysosomes are essential for optimal functioning of astrocytes and microglia (Kreher, 2021).
For more information on how impaired microglial autophagy leads to neurodegenerative diseases, please refer to: Impaired Microglial Autophagy in Neurodegenerative Diseases.
See also: Autophagy, Parkinson's Disease, and Dopaminergic Neurons
How does lysosomal dysfunction contribute to AD, PD, and ALS?
Lysosomal dysfunction can manifest in a multitude of ways. Dysfunctions in lysosomal acidification, lysosomal hydrolase activity, permeability of the lysosome membrane, and disrupted fusion of autophagosomes or endosomes with the lysosome are commonly found in neurodegenerative diseases (Kreher, 2021; Quick, 2023). These lysosomal dysfunctions result in the inefficient degradation of cellular debris that can lead to further pathogenesis.
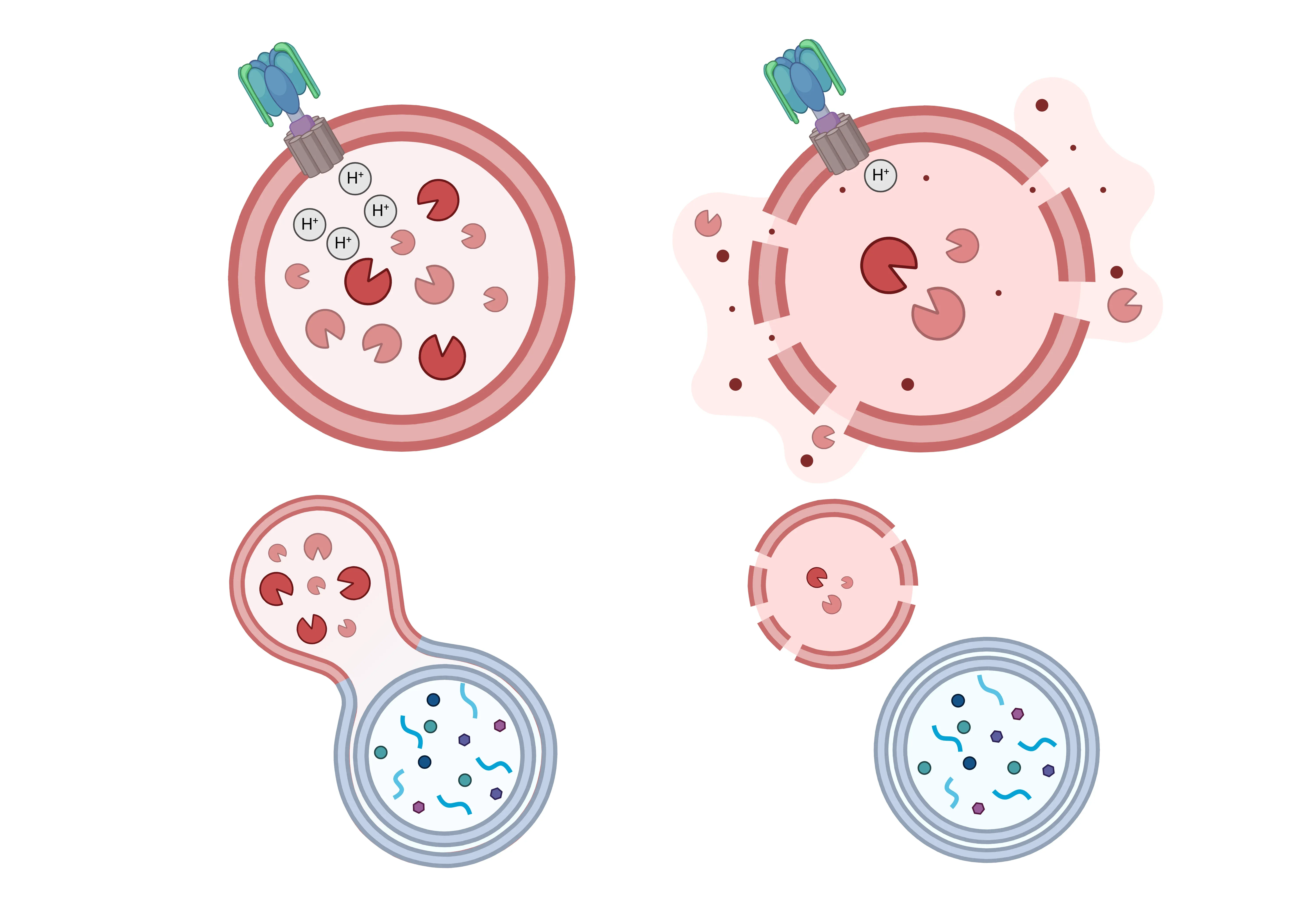
Lysosomal Dysfunction in Neurodegeneration.
Healthy lysosomal function (left) is disrupted in many neurodegenerative diseases, leading to various forms of dysfunction (right). These include impaired acidification due to v-ATPase deficits, membrane permeabilization, reduced levels of lysosomal enzymes, and impaired fusion with endosomes and autophagosomes - resulting in the buildup of undegraded materials.
 Click to copy link
Click to copy link
Alzheimer’s Disease (AD)
AD is a neurodegenerative disease marked by the accumulation of amyloid-β (Aβ) and tau aggregates. Lysosomes in astrocytes and microglia play an important role in the internalization and degradation of these toxic protein aggregates (Kreher, 2021). However, phagocytosis and degradation of Aβ appear to be more prominent in the microglia (Prakash, 2021). Mice with autophagy-deficient microglia show a reduction in available lysosomal enzymes and impaired clearance of Aβ (Choi, 2023). In addition, there is a loss of lysosomal enzymes in AD leading to reduced capacity for degradation of protein aggregates (Kim, 2025). Proper lysosomal acidification is imperative for the lysosome to serve its catabolic function and is impaired in AD. Genetic knockdown of the ClC-7 chloride and hydrogen ion antiporter leads to improper acidification of microglial lysosomes, resulting in impaired degradation of Aβ protein aggregates (Majumdar, 2011). In addition, mutations in Presenilin 1 (PS1), which have been heavily linked to the development of AD, lead to impairments in the assembly of lysosomal v-ATPase resulting in improper lysosomal acidification, as well as inefficient degradation of Aβ by microglia (Ledo, 2021). Further, PS1 mutations have also been linked to a reduction of TFEB mRNA levels in microglia, leading to a reduction in lysosomal biogenesis and impaired autophagy (Ledo, 2021).
AD and the accumulation of protein aggregates can also further exacerbate lysosomal dysfunction in glia. For instance, the presence of Aβ aggregates affect TFEB trafficking which results in further improper lysosomal acidification and reduced clearance of protein aggregates (Guo, 2017). Prolonged Aβ aggregate exposure leads to further inhibition of microglial autophagic and lysosomal capabilities by increasing the permeability of the lysosomal membrane (Pomilio, 2020). As such, a feedforward mechanism of lysosomal dysfunction and accumulation of neurotoxic protein aggregates may result in further neurodegeneration in AD.
Parkinson’s Disease (PD)
PD is the second most common neurodegenerative disease after AD and is characterized by marked alteration of motor function as well as non-motor features. The primary neuropathological hallmark of PD is the death of dopaminergic neurons within the substantia nigra. PD is also marked by the presence of α-synuclein (α-Syn) protein aggregates which is the main component of Lewy bodies. Protein aggregates in PD are largely found in neurons, but are also located in astrocytes and microglia (Tremblay, 2019). Astrocytes and microglia may play an important role in the internalization of α-Syn secreted by neurons for degradation in a lysosomal-dependent fashion (Lee, 2010; Choi, 2020). Although microglia are the primary scavengers responsible for the uptake of α-Syn (Filippini, 2019), excessive uptake of α-Syn by astrocytes can overwhelm the lysosomal pathway leading to deposits of α-Syn specifically in astrocytes (Tremblay, 2019).
Leucine-rich repeat kinase 2 (LRRK2) and glucocerebrosidase (GBA) are two of the most common genes linked to PD (Pang, 2022), and both play important roles in lysosomal functions in astrocytes and microglia (Tremblay, 2019). In astrocytes, LRRK2 is found to be co-localized with lysosomal associated membrane proteins (LAMP), which are important for facilitating lysosomal fusion with endosomes and autophagosomes for degradation (Henry, 2015; Tremblay, 2019). Mutations in LRRK2, which have been linked to PD, exhibit astrocytes with enlarged lysosomes and reduced degradative capacity (Henry, 2015). LRRK2 has been shown to negatively regulate lysosomal degradation in astrocytes and microglia potentially via its effects on TFEB (Henry, 2015; Yadavalli, 2023). Higher LRRK2 activity decreases lysosomal number in astrocytes and reduce the expression of lysosomal hydrolases in microglia (Henry, 2015; Yadavalli, 2023). The GBA gene encodes the lysosomal enzyme glucocerebrosidase (GCase), which has also been commonly linked with PD. GBA is involved in mobilization of astrocytes and microglia in response to neurodegeneration (Tremblay, 2019). In addition, knockdown of GBA reduces GCase activity and results in lysosomal dysfunction, which can increase the accumulation of α-Syn aggregates (Schöndorf, 2014). Therefore, the two genes most associated with PD appear to impact lysosomal function within astrocytes and microglia.
Amyotrophic Lateral Sclerosis (ALS)
ALS is a fatal disease characterized by the degeneration of motor neurons in the CNS. It has been discovered that a hexanucleotide repeat expansion in the non-coding region of the chromosome 9 open reading frame 72 gene (C9orf72) is one of the main underlying genetic causes of the disease (Root, 2021). Interestingly, mutations in the C9orf72 gene have also been shown to play an important role in lysosomal function. C9orf72 proteins are localized in the lysosome (Amick, 2016; Laflamme, 2019) and are highly expressed in microglia (O’Rourke, 2016). C9orf72 mutations can disrupt lysosomal acidification leading to impaired degradation (Shao, 2019). In addition, C9orf72 knockout mice show impaired lysosomal trafficking for degradation specifically in the microglia (O’Rourke, 2016). Dysfunction in the lysosomal degradation pathway induced by the C9orf72 mutations can lead to the build-up of protein aggregates that contribute to neurodegeneration in ALS (Root, 2021).
Transactive response DNA binding protein 43 kDa (TDP-43) is a protein encoded by the TARDBP gene that has also been linked to ALS (Root, 2021). TDP-43 is important for the normal functioning of lysosomes and the lysosomal-degradation pathway. For instance, TDP-43 mediates the expression of TFEB responsible for regulating lysosomal function and biogenesis (Root, 2021). In addition, mutations of TDP-43 are linked with lysosomal dysfunction (Kreiter, 2018). Interestingly, loss of TDP-43 in microglia has been shown to enhance lysosomal biogenesis, promoting microglial phagocytosis and protein aggregate clearance (Paolicelli, 2017). TDP-43 aggregates can also accumulate in neurons and glia in many neurodegenerative diseases, including ALS (Root, 2021). Excess TDP-43 can disrupt the degradation capabilities of lysosome, which can trigger aberrant autophagy as well as reduce capacity to clear TDP-43 aggregates (Leibiger, 2018). This effect is highly detrimental as clearance of TDP-43 aggregates in ALS becomes dependent on the autophagy-lysosome pathway as other degradation systems become overwhelmed (Root, 2021).
How is lysosomal function targeted to treat neurodegenerative diseases?
There is strong evidence linking neurodegenerative diseases with lysosomal dysfunction. Therefore, therapeutic strategies that improve or restore lysosomal function can be leveraged to reduce the pathological symptoms of neurodegeneration. Strategies involved are often geared toward increasing the clearance of protein aggregates by restoring or improving lysosomal function. For instance, a phase 2 placebo controlled clinical trial (NCT02914366) is currently under way, testing Ambroxol, which increases activity of the lysosomal enzyme GCase and has been shown to reduce α-Syn aggregates in mice (Migdalska-Richards, 2016). Another avenue that is being explored in clinical trials is the use of LRRK2 inhibitors to correct lysosomal dysfunction in the CNS (NCT04557800 and NCT04056689). These LRRK2 inhibitors have been found to be generally safe and well tolerated in both healthy controls and patients with PD, but their efficacy remains to be seen (Jennings, 2023).
A common target for treatment of neurodegenerative diseases via the lysosome is through TFEB, which is heavily involved in lysosomal biogenesis and function (Kim, 2025). TFEB can be activated via inhibition of the mammalian target of rapamycin (mTOR) pathway, which is achieved by molecules such as rapamycin (Kim, 2025). Other molecules can also increase TFEB in a mTOR-independent pathway and have been shown to increase degradation of Aβ protein aggregates (Portbury, 2017). Additionally, TFEB activation can be achieved through inhibition of other upstream processes. For instance, selective inhibition of the lipid kinase PIKfyve with AIT-101 activates TFEB and facilitates lysosomal clearance of TDP-43 aggregates and reduces neuroinflammation and functional deficits in a mouse model of ALS (Young, 2023). Further development of molecules that activate TFEB and facilitate lysosomal clearance, especially in glia, show great potential for the treatment of neurodegenerative diseases. Enhancing the expression of TFEB and other proteins involved in autophagy improve microglial activation to combat neuroinflammation and clearance of α-Syn aggregates (Chen, 2021). In addition, greater expression of TFEB also enhances lysosomal biogenesis and astrocytic uptake of Aβ and Tau aggregates (Xiao, 2014; Martini-Stoica, 2018). Restoring lysosomal function in glia may provide novel approaches in the treatment of neurodegeneration diseases.
Our team would be happy to answer any questions about lysosomal dysfunction in microglia & astrocytes in neurodegenerative diseases or provide specific information about the AD, ALS, and PD models we use for therapeutic efficacy studies.
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on lysosome dysfunction in microglia & astrocytes and and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
Mitochondrial Dysfunction in Microglia & Astrocytes
The role of mitochondrial dysfunction in microglia and astrocytes in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and ALS.
Impaired Microglia Autophagy in Neurodegenerative Diseases
How impaired microglia autophagy contributes to the progression of neurodegenerative diseases.
Autophagy and Transcription Factor EB (TFEB)
An overview of Transcription Factor EB (TFEB) and its role in autophagy and neurodegenerative diseases.
Autophagy & Neurodegenerative Diseases
An overview of how cellular autophagy plays a role in brain health and neurodegeneration.
Autophagy, Parkinson's Disease, and Dopaminergic Neurons
An overview of how impaired autophagy can lead to pathologic changes and neurodegeneration in dopaminergic neurons in Parkinson’s disease.