A Guide to ALS Models for Drug Discovery
A Resource for the most effective use of research animal models (mouse & rat models) of Amyotrophic Lateral Sclerosis (ALS) for preclinical testing of therapeutics.
What constitutes a good research model of ALS for drug development?
The efforts to develop new, effective therapeutics for Amyotrophic Lateral Sclerosis (ALS) (the most common form of Motor Neuron Disease [MND]) have accelerated in recent years. A broad range of targets and mechanisms of action are actively being pursued. Given that in vivo testing in animal models is an essential phase between preclinical in vitro studies and human clinical trials, it is critical that valid models that faithfully replicate multiple aspects of the human disease are used. The optimal animal model of ALS should include several key features described in the infographic below.
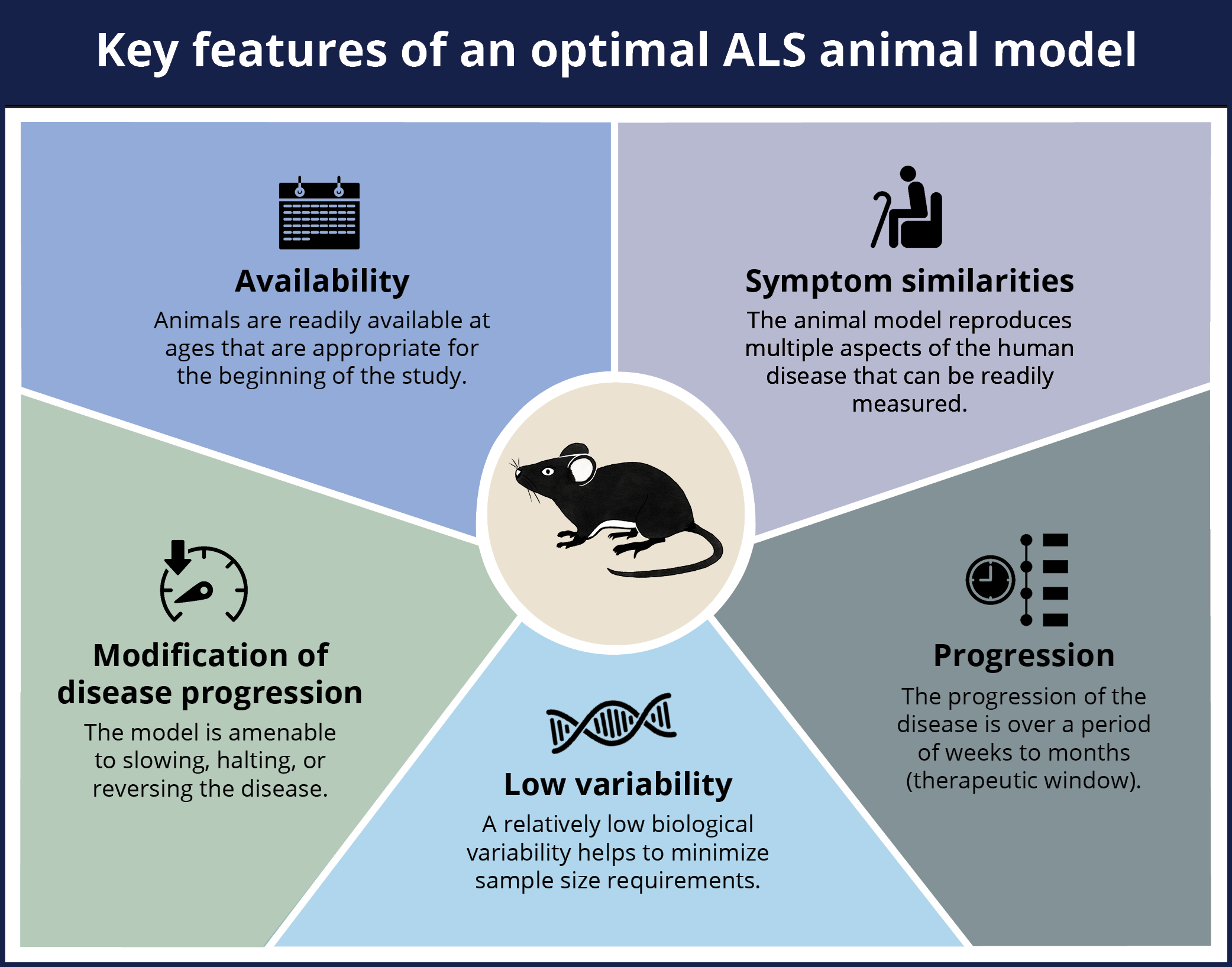
An optimal animal model of ALS should include the following five key features: availability of age-appropriate mice, symptom similarities between the model and the human disease, low variability between the animals, progression of the disease over a period of time, the ability to modify the disease progression in the model.
 Click to copy link
Click to copy link
What are key features of human ALS that should be present in an animal model?
ALS is a progressive neuromuscular disease characterized by loss of upper and lower motor neurons, resulting in muscle weakness and, ultimately, death. The disease can also include a range of non-motor features, including cognitive and behavioral alterations. A pathologic hallmark of both sporadic and familial ALS is TDP-43 proteinopathy, specifically the aggregation of this misfolded protein in the cytoplasm, thereby resulting in toxic loss and gain of function effects. A spatiotemporal pattern of TDP-43 pathology has been proposed by Braak and colleagues and is illustrated below.
Model of the spatiotemporal pattern of spread of TDP-43 pathology in the human ALS brain (adapted from Braak et al., 2013).
Like other neurodegenerative diseases, ALS is now viewed as a multi-system disease that is not limited to the central nervous system (CNS), but rather involves many parts of the body. The infographic below highlights some of systems involved in this complex disease.
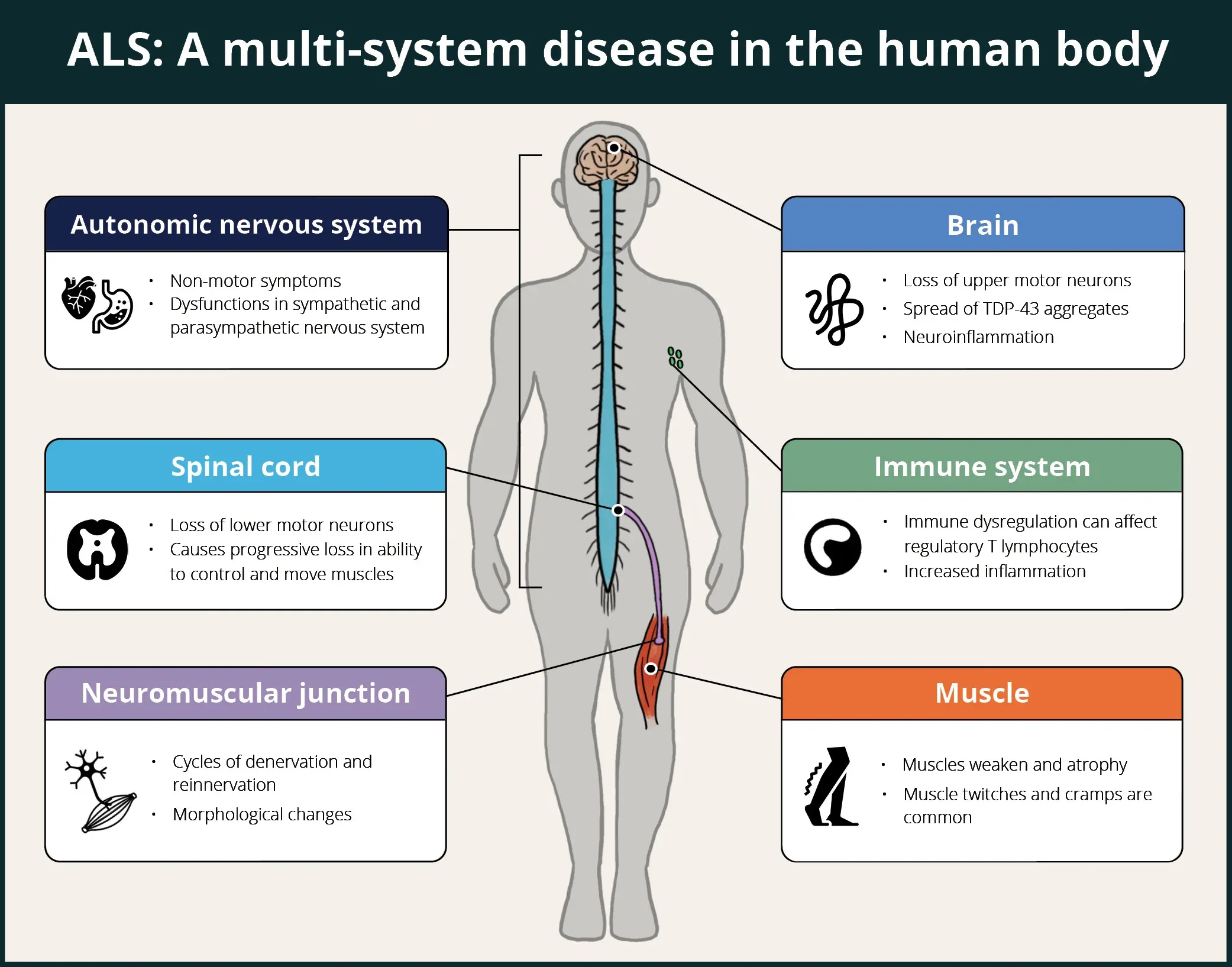
ALS is a multi-system disease in human patients affecting the brain, spinal cord, autonomic nervous system, immune system, neuromuscular junctions, and muscles.
- Click to copy link
What measures (including translational biomarkers) can be used to evaluate efficacy in ALS models?
A key element to the successful use of ALS mouse models for evaluating the efficacy of therapeutic agents is the use of robust tests that have the sensitivity required to detect disease modification. A multi-modality approach is ideal to capture how a therapy may act at the systems, cellular, and molecular levels. Additional benefit can be gained by employing "translational biomarkers" that could also be leveraged in human clinical trials.
The illustration below provides an overview of some categories of measures that have been used in efficacy studies involving ALS models. Non-invasive imaging measures, such as MRI-derived brain volumes & cortical thickness and glucose metabolism via [18F]FDG PET, are excellent translational biomarkers given that they can be utilized effectively in preclinical animal models studies and human clinical trials. A range of motor function tests to assess general locomotion (e.g. open field), balance & coordination (e.g. rotarod, beam traversal), gait (e.g. CatWalk), muscle strength (e.g. wire hang, grip strength), hindlimb clasping, tremor, and paralysis can be employed to evaluate modification of the clinical phenotype in mouse models. Muscle electrophysiology biomarkers are also clinically translational, where electromyography (EMG) is a standard assessment in ALS patients. In mouse models of ALS, Compound Muscle Action Potential (CMAP) and Motor Unit Number Estimation (MUNE) can be readily measured from the mouse hindlimb muscles (e.g. gastrocnemius). Neurofilament light has become a widely used fluid-based biomarker in ALS studies, and can also be used as a measure of axonal degeneration/injury & neurodegeneration in ALS mouse models. Finally, a wide range of techniques, such as immunophenotyping of inflammatory cells via flow cytometry, and multiplex immunofluorescence of protein expression coupled with spatial biology analysis, are utilized to assess cellular & molecular changes resulting from therapeutic intervention.
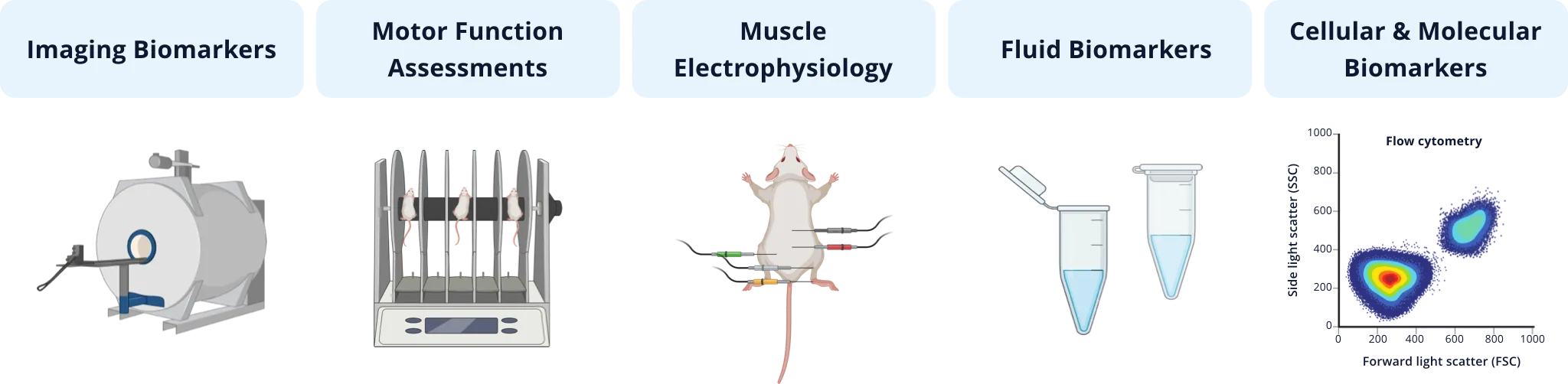
Types of quantitative measures that have been used in ALS preclinical therapeutic efficacy studies involving rodent models.
As an illustrative example of this multi-modality strategy, our team at Biospective has validated a set of these measures in the TDP-43 ΔNLS (rNLS8) transgenic mouse model of ALS, and we are actively evaluating a number of other markers that have been used in human studies. We typically use a combination of these measures, often depending on the target(s) and mechanism(s)-of-action of the therapeutic agent under study to evaluate efficacy. As an example of a translational biomarker, we routinely evaluate brain atrophy in this model using MRI, which shows progressively reduced volume and cortical thickness in the motor cortex, similar to findings in human studies.
Discover more about our ALS Models
Related Content
Up-to-date information on Amyotrophic Lateral Sclerosis and best practices related to the evaluation of therapeutic agents in ALS animal models.
Neuromuscular Junction (NMJ) Morphology & ALS Models
Insights into neuromuscular junction (NMJ), its role in amyotrophic lateral sclerosis (ALS), and tools & methods used to study morphological changes in NMJs.
ALS Mouse Models & Spinal Motor Neurons
An overview of the involvement of spinal motor neurons in disease progression in mouse models of Amyotrophic Lateral Sclerosis (ALS).
TDP-43 ΔNLS (rNLS8) Mice for ALS Drug Development
This resource provides information about the use of the ΔNLS (deltaNLS, hTDP-43ΔNLS, hTDP-43DeltaNLS, dNLS, TDP43 NLS, rNLS8) TDP-43 transgenic mouse model of ALS for preclinical therapeutic studies.
Microglia Morphology in ALS, Alzheimer's Disease & Parkinson's Disease
An overview of microglial morphological analysis and the applications to neurodegenerative disease research and drug discovery & development.
Brain Atrophy Analysis in Mouse Models of Neurodegeneration
Automated in vivo MRI-based quantitative brain atrophy measures (regional brain volumes and cortical thickness) from mouse models of ALS & Parkinson's disease.