Preformed Fibrils – A Guide to Cell and Animal Models
An overview of preformed fibril-induced cell & animal models for preclinical testing of disease-modifying therapies across multiple neurodegenerative diseases.
What are pre-formed fibrils?
Protein aggregation represents a central pathological hallmark of a wide range of neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), progressive supranuclear palsy (PSP), corticobasal degeneration, multiple system atrophy (MSA), and Huntington’s disease (HD).
Under pathological conditions, proteins such as tau, amyloid-beta, alpha-synuclein, TDP-43, and huntingtin, misfold from their native states and assemble into toxic oligomeric and fibrillar structures (Wilson, 2023). This misfolding occurs through a process of nucleated polymerization or “seeding,” whereby aggregates grow through the stepwise addition of monomers.
Once the cluster expands beyond a critical size – ranging from small soluble oligomers to highly ordered, β-sheet-rich fibrillar aggregates – further monomer addition becomes energetically favorable (Koga, 2021). This process drives the formation of toxic aggregates that are the primary components of pathological inclusions found in affected neurons and glial cells in multiple neurodegenerative disorders.
Among these assemblies, the fibrillar form represents a particularly dynamic and pathogenic state of protein aggregation. Fibrils undergo rapid elongation by recruiting soluble monomeric proteins, serving as templates that drive further aggregation (Powers, 2006). Importantly, mature fibrils are not static end-products; instead, they can spontaneously fragment into shorter fibrillar fragments that retain the ability to act as “seeds” capable of inducing de novo aggregation of endogenous proteins within recipient cells (Powers, 2007). These seeds can disseminate between cells via various mechanisms, including:
- Exocytosis
- Endocytosis
- Tunneling nanotubes
- Synaptic transmission
This self-propagating, prion-like behavior is now recognized as a unifying mechanism underlying the progressive spread of pathology across interconnected brain regions in a wide range of neurodegenerative diseases (Yamaguchi, 2017; Gibbons, 2019).
Pre-formed fibrils (PFFs) are in vitro generated fibrillar assemblies capable of recapitulating the structural and biochemical properties of disease-associated aggregates in cell and animal models. PFFs possess potent seeding activity, enabling them to recruit endogenous soluble proteins and induce intracellular aggregate formation when introduced into biological systems (Uemura, 2025).
Consequently, PFF-based models reproduce key aspects of human neurodegenerative pathology, including the misfolding, seeding, aggregation, and propagation of disease-relevant proteins, as well as downstream neurotoxicity and neuroinflammation (Majid, 2023).
The development of accurate, reliable, and physiologically relevant disease models is essential for elucidating the molecular mechanisms driving neurodegeneration, assessing the efficacy of therapeutic interventions, and evaluating the safety of drug candidates. Compared with traditional methods that rely on genetic overexpression, chemical induction, or physical injury, PFF-induced models offer distinct advantages, including:
- More rapid induction of neurodegenerative processes compared to transgenic animal models, which require long maturation periods.
- More faithful recapitulation of the naturally occurring seeding and spreading processes observed in human disease.
- Controlled induction of pathology with defined temporal and spatial characteristics.
- Less costly to establish and maintain compared to generating and breeding transgenic animals.
- Formation of PFFs can be standardized and verified (e.g. via Thioflavin T assays, Transmission Electron Microscopy, or Atomic Force Microscopy), allowing consistent quality and structure.
- PFF-induction can be combined with transgenic or toxin-induced models to better reproduce both molecular and behavioral aspects of disease.
- Useful in both in vitro and in vivo systems, from cultured neurons and iPSC-derived cells to animal studies.
Therefore, PFF-based modeling represents a powerful and innovative strategy for studying the initiation, propagation, and consequences of protein aggregation in neurodegenerative diseases (Stroo, 2017).
|
Feature |
Tau PFFs |
Amyloid-β PFFs |
Alpha-synuclein PFFs |
TDP-43 PFFs |
Huntingtin PFFs |
|
Protein type | Microtubule-associated protein | Small peptide (Aβ 40–42 aa) | Presynaptic neuronal protein | RNA-binding protein | PolyQ-expanded protein |
|
Disease modeled
|
AD, FTD, other tauopathies | Alzheimer’s disease | PD, Multiple System Atrophy, Dementia with Lewy Bodies | ALS & FTD |
Huntington’s disease |
|
Protein
| Various isoforms (3R/4R), mutations (P301S, P301L) | Mainly Aβ40/42 peptides | Wild-type and familial mutants (A53T) | Wild-type and pathological mutants | PolyQ expansions in exon 1 |
|
Fibril morphology
| Paired helical filaments (PHFs), straight filaments | Amyloid plaques/fibrils | Lewy body-like fibrils | Cytoplasmic inclusions | Nuclear and cytoplasmic inclusion fibrils |
|
Seeding efficiency
| Moderate, strain-dependent | High, rapid Aβ aggregation | High, size-dependent | Moderate to low | Variable, depends on polyQ length |
|
Cellular targets
| Neurons, axons | Extracellular plaques, neuronal synapses, neurons | Neuronal synapses, neurons | Neurons and glia | Neurons, nuclear and cytoplasmic compartments |
This table compares features used to model various neurodegenerative diseases, including protein type, disease modeled, protein isoforms/mutations, fibril morphology, seeding efficiency, and cellular targets.
Which cell models are used to study PFF-induced seeding & spread?
Cell models of PFFs commonly refer to in vitro cellular systems where PFFs are used to induce aggregation of endogenous proteins, modeling the pathology of neurodegenerative diseases (Volpicelli-Daley, 2014). The formation and propagation of PFFs in cell models usually occurs through the following steps:
- Recombinant monomers are first assembled into fibrils that structurally resemble those found in pathological inclusions.
- The PFFs are then sonicated into shorter fragments and applied to cultured cells.
- Inside cultured neurons and glia, the PFFs seed the aggregation of the cell’s natively aggregated protein.
- Depending on the PFF and the pathology it aims to mimic, this aggregation may then lead to post-translational modifications (such as phosphorylation) and formation of pathogenic inclusions similar to those seen in disease states (Stroo, 2017).
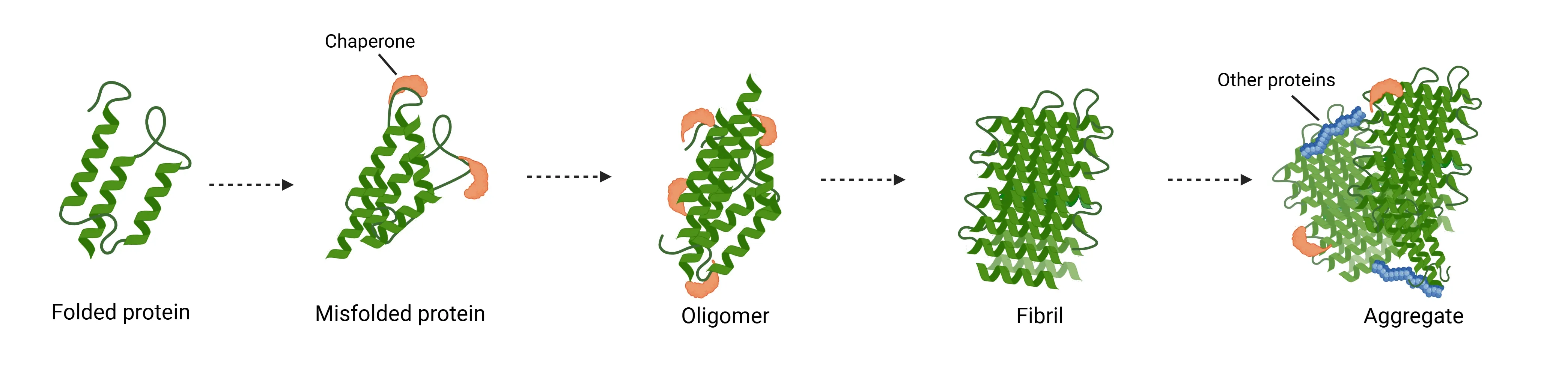
Mechanism of protein aggregation in neurodegenerative diseases.
In Parkinson's disease, for example, PFF cell models are widely used because they do not rely on overexpression of alpha-synuclein; instead, they seed aggregation at physiological protein levels, making them closer to the human condition (Benskey, 2016; Uemura, 2025).
These models allow for the study of mechanisms of alpha-synuclein aggregation, propagation, and associated cellular dysfunctions. Common cell lines used include:
- SH-SY5Y human neuroblastoma cells
- Primary neurons isolated from rodents
- Human iPSC-derived neurons
The pathological aggregates formed in these models are stable and can be propagated through cell passages, reproducing key features of synucleinopathy.
Currently, α-synuclein PFFs (including both wild-type and mutant forms) derived from either mouse or human proteins are generated under various buffer conditions. Human α-synuclein fibrils can also be produced by “seeding” reactions, in which PD patient samples are added to promote amplification of the pathogenic conformation (Wu, 2024). These models have been instrumental in investigating cellular uptake pathways, inflammatory responses, and testing aggregation inhibitors. They are also widely used in cellular and animal models to study PD mechanisms and support global drug discovery efforts.
However, variability in the preparation of PFFs or sonication parameters can affect the pathogenicity of the fibrils, which is a limitation of the system. Despite this potential limitation, PFF-based cell models remain a powerful tool for studying alpha-synuclein misfolding and its cellular consequences in neurodegenerative disease research (Dovonou, 2023).
To assess PFF-induced seeding and spread, several measures are used to evaluate the mechanisms, kinetics, and modulators of pathological fibril propagation in disease-relevant cell models.
|
Experimental Readout |
Description |
Primary Detection Methods |
|
Intracellular Aggregate Formation |
Detects newly formed intracellular aggregates (e.g. phosphorylated tau, pSer129-α-synuclein) |
Immunohistochemistry (IHC), Immunofluorescence (IF), ELISA-based aggregation-induced emission assays |
|
Fibril Uptake and Internalization |
Monitors internalization of PFFs and early seeding events |
IF microscopy, biochemical fractionation, Western blot |
|
Cell Viability |
Assesses cytotoxic effects of PFF treatment on cell health. |
MTT assay, LDH release assay, Live/Dead cell staining |
|
Seeding Capacity |
Measures ability of lysates or media from treated cells to induce fibril formation in monomeric substrates. |
Thioflavin T (ThT) fluorescence assay, Seeding ELISA, Filter trap assay |
|
Cell-to-Cell Spread |
Evaluates transmission of aggregates from donor to naïve cells. |
Co-culture assay, Conditioned media transfer, Fluorescence microscopy |
|
Pathological Co-localization |
Examines overlap between aggregates and markers of autophagy, ubiquitination, or stress pathways. |
Immunofluorescence co-staining, Confocal microscopy, Western blot |
|
Confocal Imaging |
Quantifies aggregate formation and spatial distribution within cells or cultures. |
Confocal laser scanning microscopy |
This table compares experimental readouts for assessing PFF-induced protein aggregation, propagation, and toxicity, including intracellular aggregate formation, fibril uptake and fibrillization, cell viability, seeding capacity, cell-to-cell spread, pathological co-localization, and confocal imaging.
Which animal models are used to model PFF-induced seeding & spread?
In animal models of neurodegenerative disease, PFFs are often used to generate more physiologically accurate models of pathology by mimicking the natural processes of protein misfolding, seeding, and spreading that occur in disease (Gibbons, 2019).
Generated in vitro from pathogenic proteins such as tau or alpha-synuclein, PFFs replicate critical hallmarks of neurodegeneration in vivo including:
- More accurate seeding and spread of pathogenic protein aggregates in the brain.
- Faster and more reproducible induction of molecular pathology compared with traditional transgenic or toxin-induced animal models.
- More precise control over the timing, location, and level of pathology initiation.
- More versatile and cost-effective approach applicable to multiple proteinopathies.
- Provides a robust platform for testing candidate therapies aimed at inhibiting aggregation, spread, or downstream neurodegeneration in vivo.
Overall, the use of PFFs in animal models offers a powerful and flexible approach to faithfully replicate molecular and pathological aspects of neurodegenerative diseases with temporal and spatial control, facilitating mechanistic research and therapeutic development.
Animal models used to study PFF-induced seeding and spread primarily involve rodents, especially mice and rats, with the following key models and features:
Transgenic Mice Overexpressing Human Proteins:
- These mice develop accelerated proteinopathies and behavioral or motor deficits after injection of PFFs. Commonly used models include:
- M83 mice overexpressing human alpha-synuclein with the A53T mutation for PD models.
- PS19 mice expressing human Tau with the P301S mutation for tauopathy models.
- APP/PS1 mice expressing a chimeric mouse/human amyloid precursor protein (Mo/HuAPP695swe) and a mutant human presenilin 1 (PS1-dE9), both directed to neurons for AD models.
- Transgenic mice expressing human mutant huntingtin are also used for Huntington’s disease modeling.
Wild-Type (Non-transgenic) Rodents:
Wild-type mice or rats can also be injected with recombinant PFFs (alpha-synuclein, tau, amyloid-beta) to induce endogenous protein aggregation and pathology without transgene overexpression. This model enables the study of normal physiological protein roles in seeding and propagation.
At Biospective, our stereotaxic delivery of sonicated, recombinant human tau PFFs or brain extracts into the hippocampus and overlying cortex of PS19 mice helps accelerate and synchronize the onset of tau pathology in this model, compared to the spontaneous, age-dependent development seen in PS19 mice without injection.
As a preclinical neuroscience CRO, we also offer α-synuclein PFF mouse models of Parkinson’s disease, using stereotaxic inoculation of recombinant human α-syn PFFs into M83 transgenic mice or murine α-synuclein into wild-type (B6-C3H) mice.
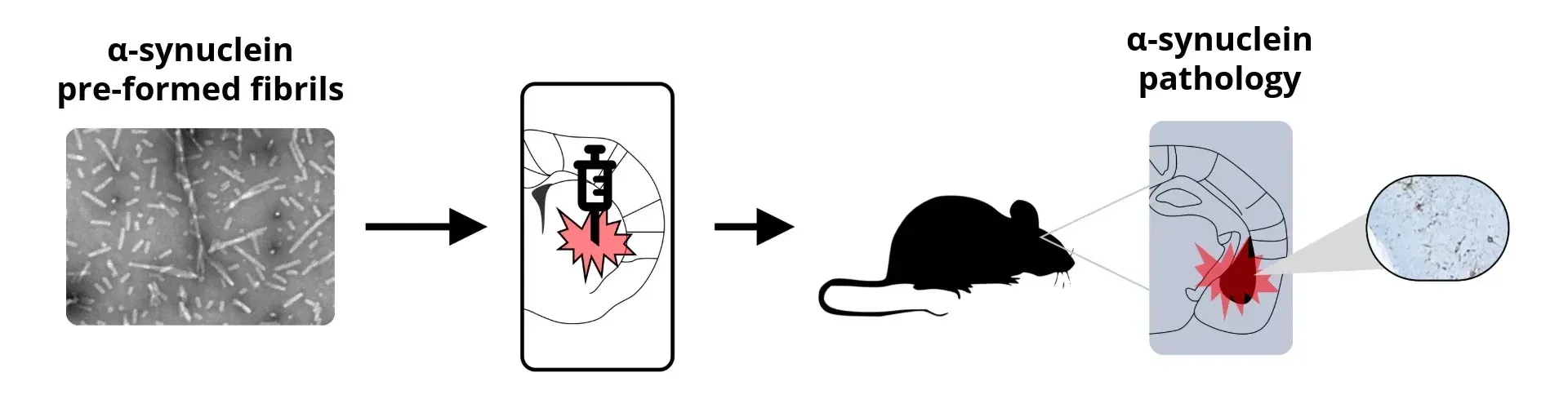
General schema for synuclein PFF animal model generation.
These animal models are highly reproducible with a nearly 100% success rate of tau and α-synuclein PFF seeding. The efficacy of PFF seeding and spread can also be validated using various outcome measures in vivo, including:
- Histopathological Analysis: Immunohistochemistry or multiplex immunofluorescence for seeded protein aggregates and disease-associated post-translational modifications in brain regions over time.
- Biochemical Analysis: Extraction and Western blot or ELISA quantification of insoluble aggregated protein fractions.
- Spatial and Temporal Spread: Mapping pathology spread along neuroanatomical pathways post PFF injection to evaluate propagation.
- Behavioral Phenotyping: Assessments of motor, cognitive, and sensory functions relevant to modeled disease to correlate pathology with functional deficits.
- Neurodegeneration and Brain Atrophy: Quantification of neuronal loss and brain atrophy via volumetric magnetic resonance imaging (MRI) or staining for synaptic markers in affected brain regions.
- In vivo Imaging: Use of reporter animals or imaging tracers to monitor aggregate formation and spread longitudinally.
- Fluid Biomarkers: Elevated levels of biomarkers of neurodegeneration – such as Neurofilament light chain (NfL) and pro-inflammatory cytokines (IL-1β, and TNF-α) – have been detected in the plasma and cerebrospinal fluid (CSF) of PFF mouse models. These biomarkers reflect cell damage associated with PFF-induced pathology.
Learn more about our characterization of various PFF-induced models of neurodegenerative diseases, our validated measures, and our Preclinical Neuroscience CRO services.
Discover more about our Neurodegenerative Diseases Models
Related Content
Up-to-date information on Neuroinflammation and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
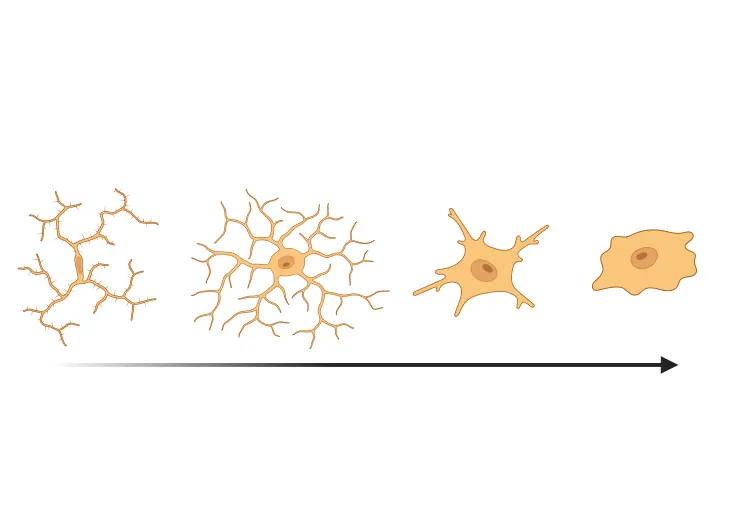
Microglial Activation in an α-Synuclein PFF Mouse Model
We have quantified microglial activation, based on morphology, in an α-synuclein preformed fibril (PFF) seeding & spreading mouse model of Parkinson’s disease.
Neurofilament Light Chain in Parkinson's Disease Models
How neurofilament light chain (NfL; NF-L) levels can be used as blood (plasma; serum) & CSF biomarkers in Parkinson's disease mouse and rat models.
Frontotemporal Dementia (FTD) & MRI Brain Atrophy
Neuroimaging biomarkers (including MRI brain atrophy) from the FTLDNI natural history study of Frontotemporal Dementia (FTD).