Fibrilles préformées – Guide des modèles cellulaires et animaux
Présentation des modèles cellulaires et animauxinduits par des fibrilles préformées pour les essais précliniques de traitements modificateurs de la maladie dans plusieurs maladies neurodégénératives.
Que sont les fibrilles préformées ?
L'agrégation des protéines est une caractéristique pathologique centrale d'un large éventail de maladies neurodégénératives, notamment la maladie d'Alzheimer (MA), la maladie de Parkinson (MP), la démence frontotemporale (DFT), la sclérose latérale amyotrophique (SLA), la paralysie supranucléaire progressive (PSP), la dégénérescence corticobasale, l'atrophie multisystématisée (AMS) et la maladie de Huntington (MH).
Dans des conditions pathologiques, des protéines telles que la protéine tau, la bêta-amyloïde, l'alpha-synucléine, la TDP-43 et la huntingtine se replient de manière anormale par rapport à leur état natif et s'assemblent en structures oligomériques et fibrillaires toxiques (Wilson, 2023). Ce repliement anormal se produit par un processus de polymérisation nucléée ou « ensemencement », dans lequel les agrégats se développent par l'ajout progressif de monomères.
Une fois que l'agrégat dépasse une taille critique – allant de petits oligomères solubles à des agrégats fibrillaires hautement ordonnés et riches en feuillets β – l'ajout de monomères supplémentaires devient énergétiquement favorable (Koga, 2021). Ce processus entraîne la formation d'agrégats toxiques qui sont les principaux composants des inclusions pathologiques présentes dans les neurones et les cellules gliales affectés dans de nombreux troubles neurodégénératifs.
Parmi ces assemblages, la forme fibrillaire représente un état particulièrement dynamique et pathogène de l'agrégation des protéines. Les fibrilles subissent un allongement rapide en recrutant des protéines monomères solubles, qui servent de modèles pour entraîner une agrégation supplémentaire (Powers, 2006). Il est important de noter que les fibrilles matures ne sont pas des produits finaux statiques ; elles peuvent au contraire se fragmenter spontanément en fragments fibrillaires plus courts qui conservent la capacité d'agir comme des « germes » capables d'induire une nouvelle agrégation de protéines endogènes dans les cellules réceptrices (Powers, 2007). Ces germes peuvent se disséminer entre les cellules par divers mécanismes, notamment :
- L'exocytose
- L'endocytose
- Nanotubes de tunneling
- La transmission synaptique
Ce comportement auto-propagateur, semblable à celui des prions, est désormais reconnu comme un mécanisme unificateur sous-jacent à la propagation progressive de la pathologie dans les régions cérébrales interconnectées dans un large éventail de maladies neurodégénératives (Yamaguchi, 2017; Gibbons, 2019).
Les fibrilles préformées (PFF) sont des assemblages fibrillaires générés in vitro capables de reproduire les propriétés structurelles et biochimiques des agrégats associés à la maladie dans des modèles cellulaires et animaux. Les PFF possèdent une puissante activité d'ensemencement, ce qui leur permet de recruter des protéines solubles endogènes et d'induire la formation d'agrégats intracellulaires lorsqu'elles sont introduites dans des systèmes biologiques (Uemura, 2025).
Par conséquent, les modèles basés sur les PFF reproduisent les aspects clés de la pathologie neurodégénérative humaine, notamment le mauvais repliement, l'ensemencement, l'agrégation et la propagation des protéines liées à la maladie, ainsi que la neurotoxicité et la neuroinflammation en aval (Majid, 2023).
Le développement de modèles de maladie précis, fiables et physiologiquement pertinents est essentiel pour élucider les mécanismes moléculaires à l'origine de la neurodégénérescence, évaluer l'efficacité des interventions thérapeutiques et évaluer la sécurité des candidats-médicaments. Par rapport aux méthodes traditionnelles qui reposent sur la surexpression génétique, l'induction chimique ou les lésions physiques, les modèles induits par les PFF offrent des avantages distincts, notamment :
- Une induction plus rapide des processus neurodégénératifs par rapport aux modèles animaux transgéniques, qui nécessitent de longues périodes de maturation.
- Une reproduction plus fidèle des processus naturels d'ensemencement et de propagation observés dans les maladies humaines.
- Une induction contrôlée de la pathologie avec des caractéristiques temporelles et spatiales définies.
- Un coût de mise en place et de maintenance moindre par rapport à la génération et à l'élevage d'animaux transgéniques.
- La formation des PFF peut être standardisée et vérifiée (par exemple, via des tests à la thioflavine T, la microscopie électronique à transmission ou la microscopie à force atomique), ce qui permet d'obtenir une qualité et une structure constantes.
- L'induction des PFF peut être combinée à des modèles transgéniques ou induits par des toxines afin de mieux reproduire les aspects moléculaires et comportementaux de la maladie.
- Utile dansles systèmes in vitro et in vivo , des neurones cultivés et des cellules dérivées d'iPSC aux études sur les animaux.
Par conséquent, la modélisation basée sur les PFF représente une stratégie puissante et innovante pour étudier l'initiation, la propagation et les conséquences de l'agrégation des protéines dans les maladies neurodégénératives (Stroo, 2017).
|
Caractéristique |
PFF tau |
PFF amyloïdes-β |
PFF alpha-synucléine |
PFF TDP-43 |
PFF de huntingtine |
|
Type de protéine | Protéine associée aux microtubules | Petit peptide (Aβ 40–42 aa) | Protéine neuronale présynaptique | Protéine de liaison à l'ARN | Protéine à expansion PolyQ |
|
Modèle de maladie
|
MA, DFT, autres tauopathies | Maladie d'Alzheimer | Maladie de Parkinson, atrophie multisystématisée, démence à corps de Lewy | SLA et DFT |
Maladie de Huntington |
|
Isoformes/mutationsde la protéine
| Diverses isoformes (3R/4R), mutations (P301S, P301L) | Principalement les peptides Aβ40/42 | Mutations de type sauvage et familiales (A53T) | Mutants de type sauvage et pathologiques | Expansions PolyQ dans l'exon 1 |
|
Morphologie des fibrilles
| Filaments hélicoïdaux appariés (PHF), filaments droits | Plaques/fibrilles amyloïdes | Fibrilles de type corps de Lewy | Inclusions cytoplasmiques | Fibrilles d'inclusion nucléaires et cytoplasmiques |
|
Efficacité d'ensemencement
| Modérée, dépendante de la souche | Élevée, agrégation rapide de l'Aβ | Élevée, dépendante de la taille | Modérée à faible | Variable, dépend de la longueur polyQ |
|
Cibles cellulaires
| Neurones, axones | Plaques extracellulaires, synapses neuronales, neurones | Synapses neuronales, neurones | Neurones et cellules gliales | Neurones, compartiments nucléaires et cytoplasmiques |
Ce tableau compare les caractéristiques utilisées pour modéliser diverses maladies neurodégénératives, notamment le type de protéine, la maladie modélisée, les isoformes/mutations protéiques, la morphologie des fibrilles, l'efficacité d'ensemencement et les cibles cellulaires.
Quels modèles cellulaires sont utilisés pour étudier l'ensemencement et la propagationinduits par les PFF ?
Les modèles cellulaires de PFF font généralement référence à des systèmes cellulaires in vitro dans lesquels les PFF sont utilisées pour induire l'agrégation de protéines endogènes, modélisant ainsi la pathologie des maladies neurodégénératives (Volpicelli-Daley, 2014). La formation et la propagation des PFF dans les modèles cellulaires se déroulent généralement selon les étapes suivantes :
- Les monomères recombinants sont d'abord assemblés en fibrilles dont la structure ressemble à celle des inclusions pathologiques.
- Les PFF sont ensuite soniquées en fragments plus courts et appliquées à des cellules cultivées.
- À l'intérieur des neurones et des cellules gliales cultivés, les PFF déclenchent l'agrégation des protéines naturellement agrégées de la cellule.
- En fonction du PFF et de la pathologie qu'il vise à imiter, cette agrégation peut alors conduire à des modifications post-traductionnelles (telles que la phosphorylation) et à la formation d'inclusions pathogènes similaires à celles observées dans les états pathologiques (Stroo, 2017).
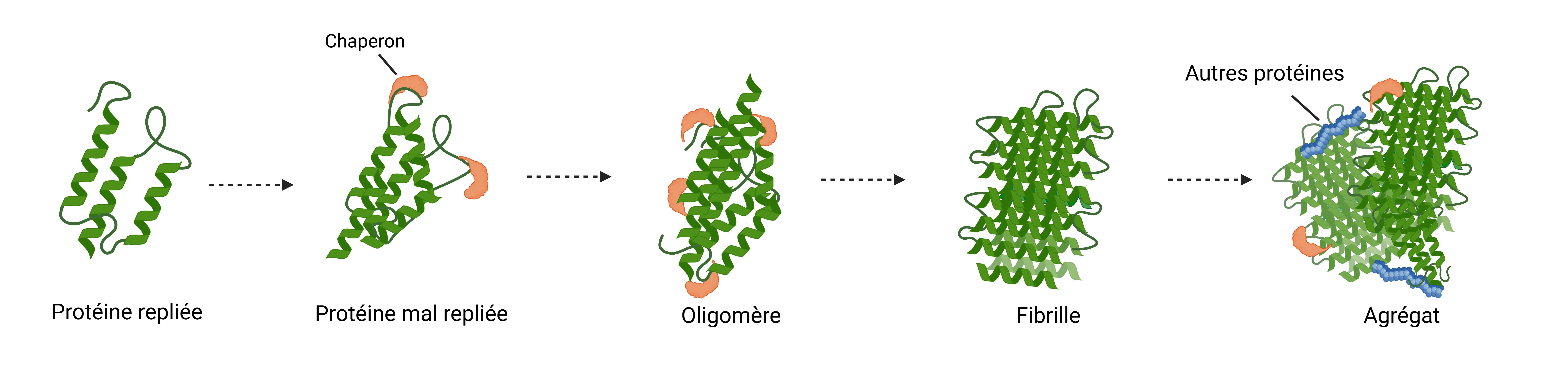
Mécanisme d'agrégation des protéines dansles maladiesneurodégénératives.
Dans le cas de la maladie de Parkinson, par exemple, les modèles cellulaires PFF sont largement utilisés car ils ne reposent pas sur la surexpression de l'alpha-synucléine ; ils favorisent plutôt l'agrégation à des niveaux physiologiques de protéines, ce qui les rend plus proches de la condition humaine (Benskey, 2016; Uemura, 2025).
Ces modèles permettent d'étudier les mécanismes d'agrégation et de propagation de l'alpha-synucléine, ainsi que les dysfonctionnements cellulaires associés. Les lignées cellulaires couramment utilisées sont les suivantes :
- Cellules de neuroblastome humain SH-SY5Y
- Neurones primaires isolés à partir de rongeurs
- Neurones dérivés d'iPSC humaines
Les agrégats pathologiques formés dans ces modèles sont stables et peuvent être propagés par passages cellulaires, reproduisant ainsi les principales caractéristiques de la synucléinopathie.
Actuellement, les PFF d'α-synucléine (y compris les formes sauvages et mutantes) dérivées de protéines de souris ou humaines sont générées dans diverses conditions tampons. Les fibrilles d'α-synucléine humaine peuvent également être produites par des réactions de « ensemencement », dans lesquelles des échantillons de patients atteints de la maladie de Parkinson sont ajoutés pour favoriser l'amplification de la conformation pathogène (Wu, 2024). Ces modèles ont joué un rôle déterminant dans l'étude des voies d'absorption cellulaire, des réponses inflammatoires et des tests d'inhibiteurs d'agrégation. Ils sont également largement utilisés dans les modèles cellulaires et animaux pour étudier les mécanismes de la maladie de Parkinson et soutenir les efforts mondiaux de découverte de médicaments.
Cependant, la variabilité dans la préparation des PFF ou les paramètres de sonication peut affecter la pathogénicité des fibrilles, ce qui constitue une limite du système. Malgré cette limitation potentielle, les modèles cellulaires basés sur les PFF restent un outil puissant pour étudier le mauvais repliement de l'alpha-synucléine et ses conséquences cellulaires dans la recherche sur les maladies neurodégénératives (Dovonou, 2023).
Pour évaluer l'ensemencement et la propagation induits par les PFF, plusieurs mesures sont utilisées afin d'évaluer les mécanismes, la cinétique et les modulateurs de la propagation pathologique des fibrilles dans des modèles cellulaires pertinents pour la maladie.
|
Lecture expérimentale |
Description |
Méthodes de détection primaires |
|
Formation d'agrégats intracellulaires |
Détecte les agrégats intracellulaires nouvellement formés (par exemple, tau phosphorylé, pSer129-α-synucléine) |
Immunohistochimie (IHC), immunofluorescence (IF), tests d'émission induite par agrégation basés sur ELISA |
|
Absorption et internalisation des fibrilles |
Surveille l'internalisation des PFF et les premiers événements de germination |
Microscopie IF, fractionnement biochimique, Western blot |
|
Viabilité cellulaire |
Évaluation des effets cytotoxiques du traitement par PFF sur la santé cellulaire. |
Test MTT, test de libération de LDH, coloration des cellules vivantes/mortes |
|
Capacité d'ensemencement |
Mesure la capacité des lysats ou des milieux provenant de cellules traitées à induire la formation de fibrilles dans des substrats monomères. |
Test de fluorescence à la thioflavine T (ThT), test ELISA de semis, test de piégeage par filtre |
|
Propagation de cellule à cellule |
Évalue la transmission des agrégats des cellules donneuses aux cellules naïves. |
Test de co-culture, transfert de milieu conditionné, microscopie à fluorescence |
|
Colocalisation pathologique |
Examine le chevauchement entre les agrégats et les marqueurs de l'autophagie, de l'ubiquitination ou des voies de stress. |
Co-coloration par immunofluorescence, microscopie confocale, Western blot |
|
Imagerie confocale |
Quantifie la formation d'agrégats et leur distribution spatiale dans les cellules ou les cultures. |
Microscopie confocale à balayage laser |
Ce tableau compare les résultats expérimentaux pour évaluer l'agrégation, la propagation et la toxicité des protéines induites par les PFF, y compris la formation d'agrégats intracellulaires, l'absorption et la fibrillation des fibrilles, la viabilité cellulaire, la capacité d'ensemencement, la propagation de cellule à cellule, la colocalisation pathologique et l'imagerie confocale.
Quels modèles animaux sont utilisés pour modéliser la propagation et la disséminationinduites par les PFF ?
Dans les modèles animaux de maladies neurodégénératives, les PFF sont souvent utilisées pour générer des modèles pathologiques plus précis sur le plan physiologique en imitant les processus naturels de mauvais repliement, de propagation et de dissémination des protéines qui se produisent dans le cadre de la maladie (Gibbons, 2019).
Générées in vitro à partir de protéines pathogènes telles que la protéine tau ou l'alpha-synucléine, les PFF reproduisent les caractéristiques essentielles de la neurodégénérescence in vivo, notamment :
- Une propagation et une dissémination plus précises des agrégats de protéines pathogènes dans le cerveau.
- Une induction plus rapide et plus reproductible de la pathologie moléculaire par rapport aux modèles animaux transgéniques ou induits par des toxines traditionnels.
- Un contrôle plus précis du moment, du lieu et du niveau d'apparition de la pathologie.
- Une approche plus polyvalente et plus rentable, applicable à plusieurs protéinopathies.
- Une plateforme robuste pour tester des thérapies candidates visant à inhiber l'agrégation, la propagation ou la neurodégénérescence en aval in vivo.
Dans l'ensemble, l'utilisation des PFF dans les modèles animaux offre une approche puissante et flexible pour reproduire fidèlement les aspects moléculaires et pathologiques des maladies neurodégénératives avec un contrôle temporel et spatial, facilitant ainsi la recherche mécanistique et le développement thérapeutique.
Les modèles animaux utilisés pour étudier l'ensemencement et la propagation induits par les PFF impliquent principalement des rongeurs, en particulier des souris et des rats, avec les modèles et caractéristiques clés suivants :
Souris transgéniques surexprimant des protéines humaines:
- Ces souris développent des protéinopathies accélérées et des déficits comportementaux ou moteurs après injection de PFF. Les modèles couramment utilisés sont les suivants :
- Les souris M83 surexprimant l'alpha-synucléine humaine avec la mutation A53T pour les modèles de la maladie de Parkinson.
- Les souris PS19 exprimant la protéine Tau humaine avec la mutation P301S pour les modèles de tauopathie.
- Les souris APP/PS1 exprimant une protéine précurseur amyloïde chimérique souris/humaine (Mo/HuAPP695swe) et une préséniline 1 humaine mutante (PS1-dE9), toutes deux dirigées vers les neurones pour les modèles de MA.
- Des souris transgéniques exprimant la huntingtine mutante humaine sont également utilisées pour la modélisation de la maladie de Huntington.
Rongeurs de type sauvage (non transgéniques) :
Les souris ou rats de type sauvage peuvent également recevoir des injections de PFF recombinants (alpha-synucléine, tau, bêta-amyloïde) afin d'induire une agrégation protéique endogène et une pathologie sans surexpression transgénique. Ce modèle permet d'étudier les rôles physiologiques normaux des protéines dans l'ensemencement et la propagation.
Chez Biospective, notre administration stéréotaxique de PFF tau humaines recombinantes soniquées ou d'extraits cérébraux dans l'hippocampe et le cortex sus-jacent de souris PS19 contribue à accélérer et à synchroniser l'apparition de la pathologie tau dans ce modèle, par rapport au développement spontané et dépendant de l'âge observé chez les souris PS19 sans injection.
En tant qu'organisme de recherche sous contrat (CRO) spécialisé en neurosciences précliniques, nous proposons également des modèles murins de la maladie de Parkinson à base de PFF d'α-synucléine, en utilisant l'inoculation stéréotaxique de PFF d'α-syn recombinante humaine dans des souris transgéniques M83 ou d'α-synucléine murine dans des souris de type sauvage (B6-C3H).
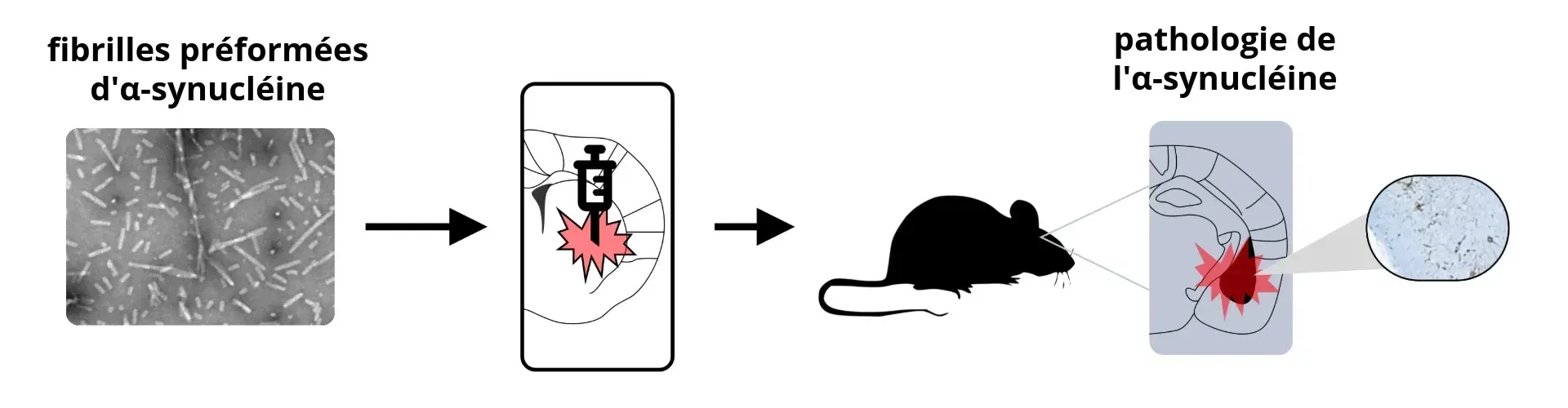
Schémagénéral pour la création d'un modèle animal PFF synucléine.
Ces modèles animaux sont hautement reproductibles, avec un taux de réussite de près de 100 % pour l'ensemencement des PFF de tau et d'α-synucléine. L'efficacité de l'ensemencement et de la propagation des PFF peut également être validée à l'aide de diverses mesures de résultats in vivo, notamment :
- Analyse histopathologique: immunohistochimie ou immunofluorescence multiplexe pour les agrégats protéiques ensemencés et les modifications post-traductionnelles associées à la maladie dans les régions du cerveau au fil du temps.
- Analyse biochimique : extraction et quantification par Western blot ou ELISA des fractions protéiques agrégées insolubles.
- Propagation spatiale et temporelle : cartographie de la propagation de la pathologie le long des voies neuroanatomiques après l'injection de PFF afin d'évaluer la propagation.
- Phénotypage comportemental: évaluations des fonctions motrices, cognitives et sensorielles pertinentes pour la maladie modélisée afin de corréler la pathologie avec les déficits fonctionnels.
- Neurodégénérescence et atrophie cérébrale : quantification de la perte neuronale et de l'atrophie cérébrale par imagerie par résonance magnétique (IRM) volumétrique ou coloration des marqueurs synaptiques dans les régions cérébrales touchées.
- Imageriein vivo: utilisation d'animaux rapporteurs ou de traceurs d'imagerie pour surveiller la formation et la propagation des agrégats de manière longitudinale.
- Biomarqueurs liquides: des niveaux élevés de biomarqueurs de neurodégénérescence, tels que la chaîne légère du neurofilament (NfL) et les cytokines pro-inflammatoires (IL-1β et TNF-α), ont été détectés dans le plasma et le liquide céphalo-rachidien (LCR) de modèles murins PFF. Ces biomarqueurs reflètent les lésions cellulaires associées à la pathologie induite par la PFF.
Veuillez découvrir notre caractérisation de divers modèles de maladies neurodégénératives induites par les PFF, nos mesures validées et nos services CRO en neurosciences précliniques.
En savoir plus sur nos modèles de maladies neurodégénératives
Contenu connexe
Informations actualisées sur la neuroinflammation et les meilleures pratiques liées à l'évaluation des agents thérapeutiques dans les modèles animaux de maladies neurodégénératives.
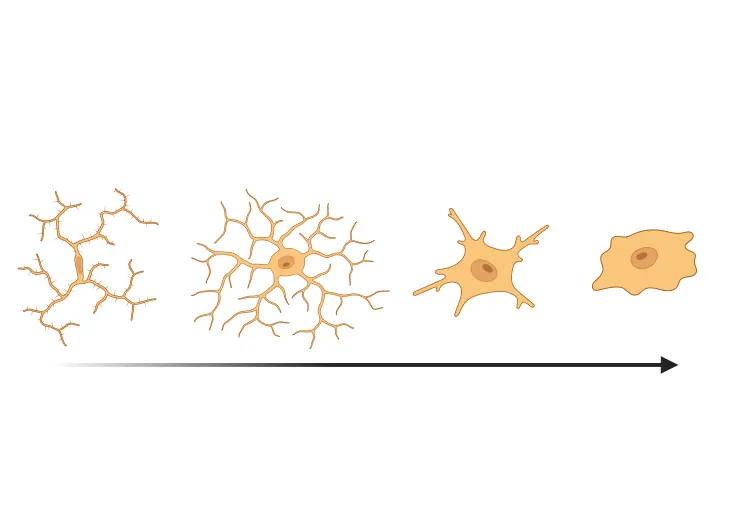
Activation microgliale dans un modèle de souris PFF à α-synucléine
Nous avons quantifié l'activation microgliale, sur la base de la morphologie, dans un modèle murin d'ensemencement et d'étalement de fibrilles préformées d'α-synucléine (PFF) de la maladie de Parkinson.
Le neurofilament à chaîne légère dans les modèles de la maladie de Parkinson
Comment les niveaux de neurofilament à chaîne légère (NfL ; NF-L) peuvent être utilisés comme biomarqueurs dans le sang et le LCR dans les modèles de souris et de rats atteints de la maladie de Parkinson.
Démence frontotemporale (DFT) et atrophie cérébrale par IRM
Biomarqueurs IRM (y compris l'atrophie cérébrale) issus des études FTLDNI sur l'histoire naturelle de la démence frontotemporale (DFT).