Mitophagy and Parkinson’s Disease
An overview of how impaired mitophagy can lead to neurodegeneration in Parkinson’s disease.
What is mitophagy?
Autophagy is a lysosomal degradative process that breaks down cellular debris, including misfolded proteins and damaged organelles, and recycles their components for the synthesis of new molecules (Gómez-Virgilio, 2022). Mitophagy, also known as mitochondrial autophagy, is a specialized form of autophagy that selectively degrades damaged and dysfunctional mitochondria to maintain energy homeostasis (Uoselis, 2023; Wang, 2023).
In dysfunctional or damaged mitochondria, the inner mitochondrial membrane becomes depolarized, leading to the accumulation of PINK1 (PTEN-induced kinase 1) on the outer membrane of damaged mitochondria (Jin, 2012; Narendra, 2012; Pickrell, 2015). PINK1 acts as a molecular sensor that recruits and activates Parkin, an E3 ubiquitin ligase, that ubiquitinates several outer mitochondrial membrane (OMM) proteins, initiating mitophagy and marking damaged mitochondria (Chan, 2011; Yoshii, 2011). These tagged mitochondria are then encapsulated by double membrane autophagosome vesicles, which transport them to lysosomes for degradation (Youle, 2011; Jin, 2012; Pickrell, 2015).
Mitophagy plays an important role in mitochondria quality control by supporting the generation of new and healthy mitochondria through biogenesis and ensuring that cells’ energy demands are being met. Mitochondria fission and fusion work in tandem with mitophagy to create a functional mitochondrial population; fission isolates damaged portions of mitochondria by fragmenting them into smaller daughter mitochondria to be marked for targeted degradation, while fusion optimizes mitochondria function by enabling mitochondria to compensate for each other’s defects and sustain cellular energy production (Twig, 2008; Ashrafi, 2013).
For in vivo and in vitro monitoring and quantification of mitophagy, scientists often use a combination of techniques. These include electron microscopy (EM) for high-resolution visualization of autophagosomes engulfing mitochondria, pH-sensitive fluorescent reporters, such as mitochondria-targeted Keima (mt-Keima), to detect mitochondria within lysosomes, and Western blotting to measure the levels of mitophagy-related proteins (Zhu, 2011; Williams, 2017; Jung, 2019).
When the inner mitochondrial membrane becomes depolarized, PINK1 accumulates on the outer mitochondrial membrane. The protein acts as a molecular sensor that recruits and activates Parkin, initiating mitophagy and marking damaged mitochondria. The tagged mitochondria are encapsulated by double membrane autophagosome vesicles, which transport them to the lysosome for degradation.
What is the relationship between impaired mitophagy and degeneration of dopaminergic neurons in PD?
Impaired mitophagy is implicated in the etiology of PD. Genetic studies have releveled that loss-of-function mutations in key mitophagy proteins—PINK1 and Parkin—induce Lewy body pathology and lead to autosomal recessive early-onset PD. The Parkin gene is responsible for as many as 50% of all familial early-onset PD cases (Kitada, 1998; Lücking, 2000; Valente, 2004; Gandhi, 2006; Samaranch, 2010).
PINK1 and Parkin act as regulators of mitochondrial fission, a process that this critical for isolating damaged mitochondrial fragments to be targeted by the mitophagy machinery (Buhlman, 2014; Ge, 2020). Mutations in these proteins lead to the accumulation of abnormally large and dysfunctional mitochondria that promote alpha-synuclein aggregation within dopaminergic neurons, a prominent hallmark of PD (Srinivasan, 2021). Additionally, loss of PINK1 and Parkin reduces the solubility of alpha-synuclein, encouraging its aggregation within dopaminergic neurons (Lonskaya, 2013). Simultaneously, mutant forms of alpha-synuclein, such as A53T, cause ubiquitin-proteasome pathway (UPS) failure in dopaminergic neurons, exacerbating alpha-synuclein aggregation (McKinnon, 2020).
Mitochondrial dysfunctional is also known to cause selective degeneration of dopaminergic neurons in PD, which have particularly high metabolic demands because of their large axonal arbors, pacemaking activity, and pumping of calcium that regulates dopamine release (Arduíno, 2011; Rademacher, 2024). When mitophagy is impaired in PD, it leads to the accumulation of structurally and functionally dysfunctional mitochondria within dopaminergic neurons. These mitochondria generate heightened levels of reactive oxygen species (ROS) due to superoxide production from electron leakage, have reduced or little ATP production, cause calcium overload, and activate apoptosis pathways—all of which contribute to the degeneration and loss of dopaminergic neurons in the midbrain (Liu, 2018; Zampese, 2020).
Postmortem brain tissue analyses of PD patients confirm the role of mitophagic failures in PD pathology. These failures include Complex I dysfunction in the substantia nigra pars compacta, increased age-dependent accumulation of mitochondrial DNA (mtDNA), dysregulated expression of mitochondrial proteins, and increased oxidative damage to these proteins (Mizuno, 1989; Bender, 2006; Grünewald, 2016; Giannoccaro, 2017). These findings highlight the role of mitophagy in the pathogenesis of PD and underscore its potential as a therapeutic target for PD-related neurodegeneration.
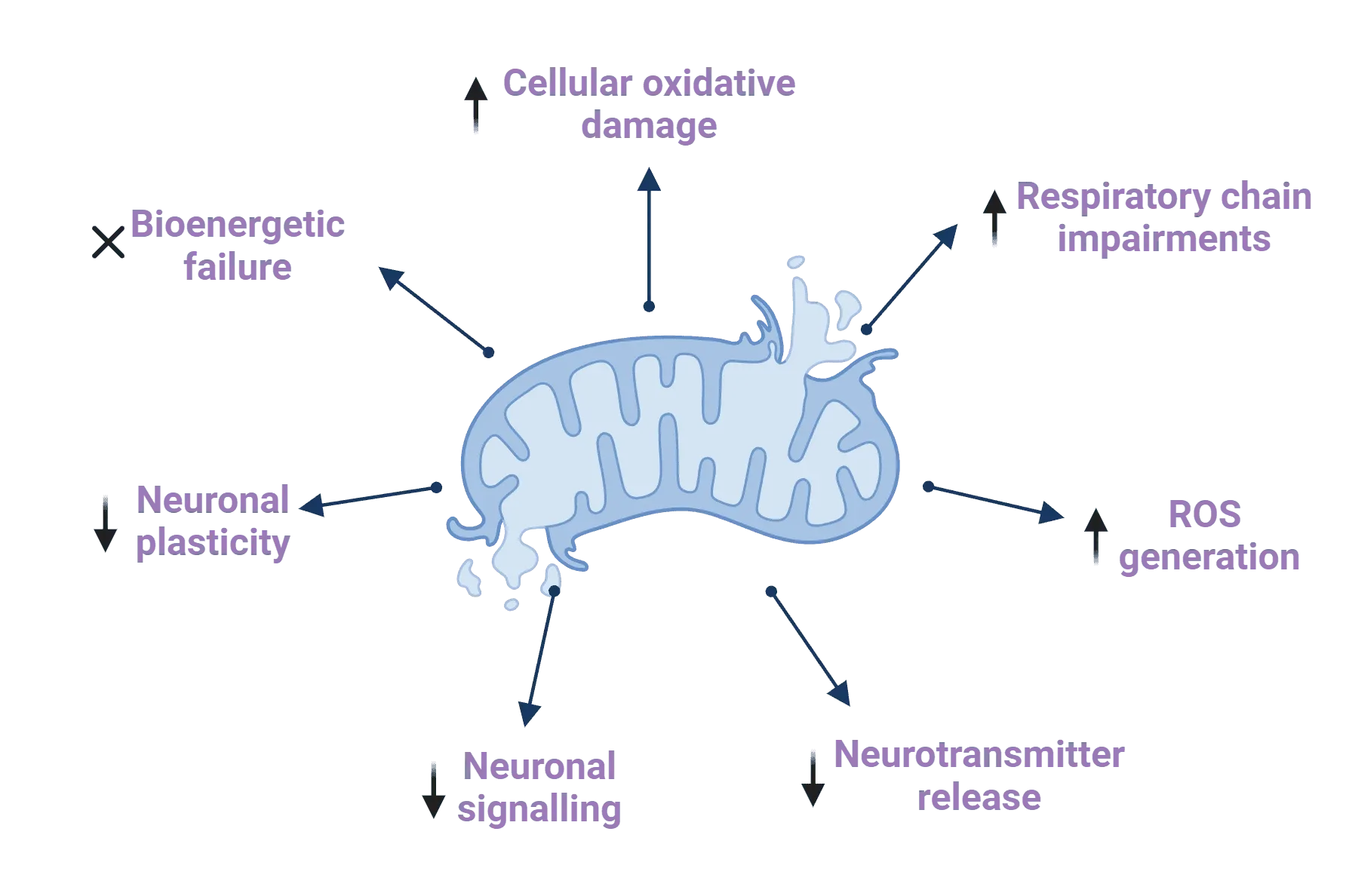
Mitochondrial dysfunction contributes to the development and progression of Parkinson's disease through a range of mechanisms. Up arrows in the infographic represent increases, while down arrows represent reductions. Figure and caption are reproduced from Millichap et al. (Millichap, 2021) under the Creative Commons Attribution License.
Have therapeutics targeting mitophagy shown efficacy in Parkinson’s disease models?
Failures in PINK1/Parkin-mediated mitophagy have been linked to the selective degeneration and death of dopaminergic neurons in PD (Ge, 2020). Hence, pharmacological regulation of PINK1/Parkin-mediated mitophagy has emerged as an attractive strategy for developing disease-modifying treatments aimed at mitigating dopaminergic neuron loss in PD.
Recent studies in PD mouse models highlight the therapeutic potential of PINK1/Parkin mitophagy activators. For example, in their latest work, Zhao et al. (Zhao, 2024) demonstrated that treatment with palmatine (PAL) improves motor function and reduces dopaminergic neuronal loss in MPTP-induced mice. PAL achieves this reduced dopaminergic neuron loss by enhancing mitophagy and inhibiting the activation of NLRP3 inflammasome, a multiprotein complex that drives inflammatory signaling. Similarly, Chen et al. (Chen, 2022) reported that dexmedetomidine protects dopaminergic neurons in MPTP-induced mice by upregulating the expression of key mitophagy proteins, thereby enhancing PINK1/Parkin-mediated mitophagy.
However, evidence from mouse and drosophila models, as well as primary neuronal cultures, indicates that loss of PINK1 and Parkin function does not consistently lead to the accumulation of dysfunctional mitochondria within cells (Goldberg, 2003; Narendra, 2011; Ge, 2020). These findings suggest the existence of alternative mitophagy pathways that may compensate for PINK1/Parkin deficiencies. Elucidating how and when these compensatory pathways are activated will offer insights into the unique vulnerability of dopaminergic neurons in PD compared to other cell populations.
Our team would be happy to answer any questions about mitophagy in neurodegenerative diseases or provide specific information about the PD models we use for therapeutic efficacy studies.
Discover more about our PD Models
Related Content
Up-to-date information on mitophagy in Parkinson's disease and best practices related to the evaluation of therapeutic agents in animal models of neurodegenerative diseases.
Mitochondrial Dysfunction in Microglia & Astrocytes
The role of mitochondrial dysfunction in microglia and astrocytes in neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and ALS.
Autophagy & Neurodegenerative Diseases
An overview of how cellular autophagy plays a role in brain health and neurodegeneration.
Autophagy, Parkinson's Disease, and Dopaminergic Neurons
An overview of how impaired autophagy can lead to pathologic changes and neurodegeneration in dopaminergic neurons in Parkinson’s disease.
Microglia Morphology in ALS, Alzheimer's Disease & Parkinson's Disease
An overview of microglial morphological analysis and the applications to neurodegenerative disease research and drug discovery & development.
Microglial Activation in an α-Synuclein PFF Mouse Model
We have quantified microglial activation, based on morphology, in an α-synuclein preformed fibril (PFF) seeding & spreading mouse model of Parkinson’s disease.
Neurofilament Light Chain in Parkinson's Disease Models
How neurofilament light chain (NfL; NF-L) levels can be used as blood (plasma; serum) & CSF biomarkers in Parkinson's disease mouse and rat models.
AAV α-Synuclein Models for Parkinson's Disease Drug Development
Overview of adeno-associated virus (AAV) induced α-synuclein expression in mouse & rat models for use in preclinical studies of disease-modifying therapeutics.