线粒体功能障碍在小胶质细胞与星形胶质细胞中的作用
线粒体功能障碍在神经退行性疾病(包括阿尔茨海默病、帕金森病和肌萎缩侧索硬化症)中的作用,涉及小胶质细胞和星形胶质细胞。
线粒体在小胶质细胞和星形胶质细胞中的作用是什么?
线粒体功能与动态
线粒体是支持多种细胞功能的必需细胞器。线粒体被称为细胞的"能量工厂",主要通过电子传递链(ETC)中的氧化磷酸化(OXPHOS)过程,利用氧气和三磷酸甘油(ATP)生成细胞活动所需的主要能量形式——三磷酸腺苷(ATP)。ETC由一系列蛋白质复合体组成(Tönnies, 2017)。然而,线粒体的功能远不止于能量代谢。线粒体还参与脂质和氨基酸代谢、钙离子(Ca²⁺)稳态以及程序性细胞死亡(PCD),常被称为凋亡(Bose, 2016)。此外,它们通过平衡活性氧(ROS)的产生与抗氧化防御系统来调节氧化应激。
线粒体具有高度动态性,通过融合和分裂过程持续进行形态重塑。这些动态变化对维持线粒体功能和防止凋亡至关重要。参与线粒体动态的关键蛋白包括负责分裂的动力蛋白相关蛋白1(Drp1),以及负责融合的线粒体融合蛋白(Mfn1/2)和视神经萎缩1(OPA1)。这些过程的平衡失调会导致线粒体碎片化、ATP生成减少及细胞应激增加。此外,分裂与融合缺陷会阻碍线粒体运输,进而引发突触功能障碍和神经退行性变。
线粒体功能障碍与胶质介导的神经炎症
线粒体功能障碍可能由多种因素引起,包括衰老和线粒体DNA(mtDNA)突变,导致ATP生成受损、活性氧(ROS)水平升高、结构异常、氧化应激和凋亡(Tönnies, 2017;Wang, 2020)。生理水平的ROS作为信号分子发挥作用,而过量ROS会破坏线粒体完整性和功能(Kumar, 2012)。未通过线粒体自噬充分清除的受损线粒体可能释放线粒体来源的损伤相关分子模式(DAMPs),通过激活先天免疫受体触发炎症信号通路(Salmina, 2021;Lin, 2022)。
小胶质细胞和星形胶质细胞在中央神经系统(CNS)免疫反应中发挥关键作用,是线粒体DAMPs的主要响应者。小胶质细胞通过模式识别受体识别这些危险信号,激活转录因子如NF-κB,进而驱动促炎细胞因子如白细胞介素-1β(IL-1β)和肿瘤坏死因子α(TNF-α)的释放。这些细胞因子加剧氧化应激,破坏血脑屏障(BBB),并促进神经元损伤(Wang, 2020)。在小胶质细胞和星形胶质细胞中,线粒体功能障碍会损害神经保护功能并维持慢性炎症。由此形成恶性循环:线粒体损伤激活胶质细胞,而产生的炎症介质进一步损伤线粒体,加剧神经退行性变(Jeon, 2020;Yan, 2020)。
值得注意的是,线粒体功能障碍可能在神经退行性疾病(如阿尔茨海默病(AD)、帕金森病(PD)和肌萎缩侧索硬化症(ALS))中先于神经炎症发生,这表明线粒体功能障碍可能是疾病病理学的潜在驱动因素(Swerdlow, 2014)。除神经元外,研究胶质细胞中的线粒体变化可能为早期疾病机制提供更全面的理解,并促进新型治疗策略的开发(Lin, 2022)。
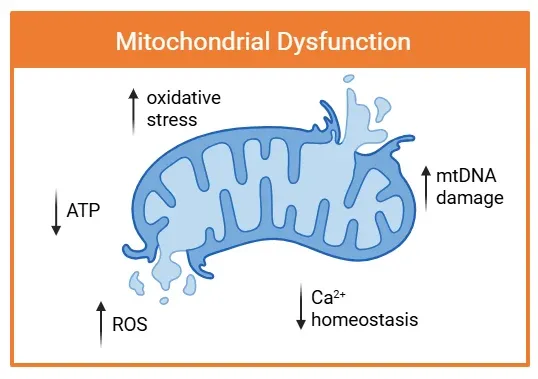
线粒体功能障碍的特征是结构损伤和功能障碍。箭头指示关键病理变化,包括活性氧(ROS)生成增加、氧化应激和线粒体DNA(mtDNA)损伤。这些功能障碍与ATP生成减少和钙离子(Ca2+)稳态紊乱同时发生,促进神经炎症和神经退行性疾病的进展。
线粒体功能障碍在小胶质细胞与星形胶质细胞中的作用及其与阿尔茨海默病(AD)、帕金森病(PD)和肌萎缩侧索硬化症(ALS)的关系是什么?
小胶质细胞和星形胶质细胞中的线粒体功能障碍是神经退行性疾病研究中一个新兴的关注领域。这些胶质细胞中线粒体功能的异常不仅会损害其支持性功能,还会引发并维持神经炎症,形成一个反馈循环,从而加剧神经元损伤。
阿尔茨海默病(AD)
AD的临床特征包括进行性记忆丧失、认知功能下降和行为改变。其病理学标志包括β-淀粉样蛋白(Aβ)斑块的积累、磷酸化tau蛋白神经原纤维缠结以及神经元死亡。值得注意的是,线粒体功能障碍、氧化应激和炎症是AD进展的早期特征之一(Swerdlow, 2014)。
在AD中,线粒体内Aβ的积累抑制电子传递链(ETC)的II和IV复合体,减少ATP生成并增加活性氧(ROS)(Kumar, 2015;Swerdlow, 2016)。Aβ还扰乱线粒体蛋白质输入、损伤线粒体DNA(mtDNA)并破坏钙离子(Ca²⁺)稳态,促进凋亡(Kumar, 2015)。此外,Aβ扰乱线粒体形态和动态,包括分裂和融合的调节(Wang, 2008;2009)。调节这些动态的Drp1和OPA1的失调与Aβ诱导的神经元毒性相关(Wang, 2008;2009)。受损线粒体的清除受阻会加剧神经退行性变(Kumar, 2015)。
前临床模型提示,靶向线粒体的干预措施可能具有治疗潜力。例如,OPA1过表达可恢复AD模型中的线粒体结构和功能(Wang, 2008;2020)。此外,P110,一种选择性Drp1/Fis1抑制剂,可减少线粒体碎裂以及小胶质细胞和星形胶质细胞的活化,减少小胶质细胞释放受损线粒体,并保护神经元免受细胞因子介导的损伤(Joshi,2019)。这些发现支持通过靶向小胶质细胞中的线粒体分裂来减轻神经炎症和神经元损伤的潜力。
帕金森病(PD)
PD表现为运动症状,包括运动迟缓、肌强直、步态障碍及静止性震颤,同时伴随认知障碍、情绪及睡眠紊乱等非运动症状。其特征为Lewy小体中α-突触核蛋白(α-syn)的积累及黑质致密部(SNpc)多巴胺能神经元的退化。尽管大多数PD病例为散发性,但部分病例与DJ-1基因(也称为PARK7)的遗传突变相关(Almikhalfi, 2020)。
无论是家族性还是散发性PD,均表现出线粒体缺陷(Grünewald, 2019)。DJ-1基因敲除大鼠在黑质中出现进行性多巴胺能神经元退化、运动功能障碍及线粒体通路紊乱,如电子传递链复合物I(ETC复合物I)(Almikhlafi, 2020)。星形胶质细胞通过自噬-溶酶体通路帮助降解α-syn;然而,过量的α-syn会压倒这一过程,导致进一步的线粒体功能障碍。α-syn与线粒体成分相互作用,包括细胞色素C氧化酶1(COX1),从而损害线粒体功能(Jeon,2020)。ATP13A2基因突变也会扰乱线粒体和溶酶体通路,促进α-syn的积累(Grünewald, 2019)。
肌萎缩侧索硬化症(ALS)
ALS是一种进行性神经退行性疾病,影响运动神经元,导致肌肉无力、痉挛和呼吸衰竭。尽管大多数病例为散发性,但家族性ALS与C9orf72 、 SOD1、 FUS和TARDBP等基因的突变相关。异常折叠的蛋白质,如突变的超氧化物歧化酶1(SOD1)和TDP-43,是ALS的标志性特征。
异常线粒体形态在ALS患者、小鼠及细胞培养模型中均有报道(Magrané, 2009)。线粒体在神经肌肉接头处的运输和定位对运动神经元功能至关重要。尸检研究显示,ALS患者脊髓中的电子传递链(ETC)复合体活性降低(Obrador, 2020)。线粒体Sirtuin蛋白(如SIRT1和SIRT3)的调控异常会损害线粒体生物发生和功能(Obrador, 2020)。SIRT1通过激活PGC-1α增强线粒体功能和ROS防御,而SIRT3参与此过程(Song, 2013)。用白藜芦醇激活SIRT1可改善SOD1G93A ALS小鼠模型的症状和生存率,而SIRT3过表达可减少ROS的产生,并保护SOD1G93A运动神经元免受线粒体碎裂和死亡(Han, 2012;Song, 2013)。
TDP-43还与线粒体蛋白相互作用,如电压依赖性阴离子通道1(VDAC1),扰乱线粒体动态和线粒体自噬(Obrador, 2020)。在诱导多能干细胞(iPSC)来源的C9orf72运动神经元中,氧化应激随年龄增加而升高,但可通过减轻氧化应激部分缓解(Lopez-Gonzalez, 2016)。
综上所述,尽管AD、PD和ALS在临床表现和病理特征上存在差异,但它们共享一个共同机制:线粒体功能障碍驱动慢性胶质细胞激活和神经炎症。这种双向关系形成反馈循环,加速疾病进展。靶向这一循环可能为治疗提供新途径。
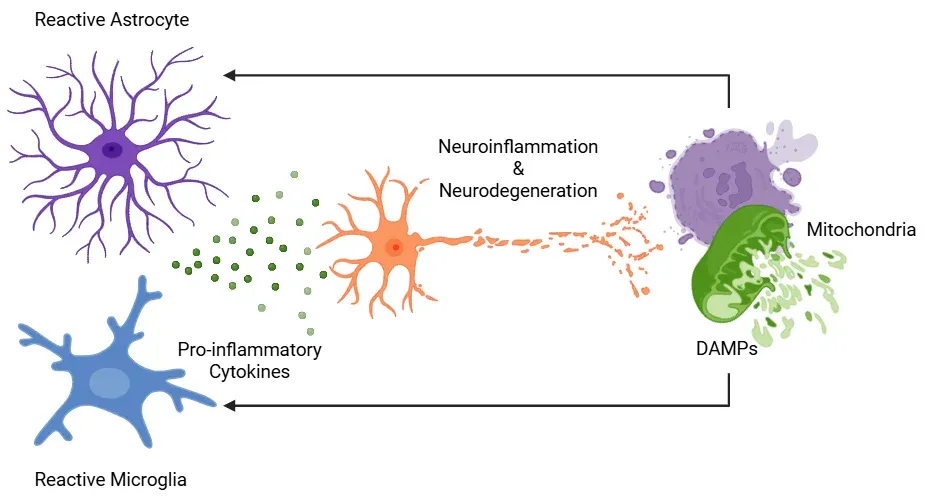
神经炎症在神经退行性疾病中是由功能障碍的线粒体与反应性胶质细胞之间的相互作用驱动的。受损的线粒体释放线粒体来源的危险相关分子模式(DAMPs),这些分子被小胶质细胞和星形胶质细胞识别。作为回应,这些胶质细胞采用反应性表型并分泌促炎性细胞因子,进一步损害线粒体功能。这一过程形成了一个自我维持的反馈循环,其中线粒体功能障碍与胶质细胞反应性相互强化,导致慢性神经炎症和神经元退行性变的进展。图改编自Picca等(Picca, 2022)在知识共享署名许可下。
神经退行性疾病中针对线粒体功能障碍的治疗方法有哪些?
鉴于线粒体功能障碍和氧化应激在神经退行性疾病中的核心作用,靶向线粒体通路已成为一种有前景的治疗策略。目前正在探索多种方法,包括线粒体靶向抗氧化剂、调节线粒体动态以及增强生物能功能。尽管存在如血脑屏障渗透等挑战,这些干预措施仍有望减缓或改变疾病进展。
氧化应激是由活性氧(ROS)与抗氧化防御系统失衡引起的,是线粒体功能障碍的关键驱动因素。线粒体靶向抗氧化剂在前临床模型中通过预防神经元氧化损伤显示出令人鼓舞的结果(Kumar, 2015)。MitoQ、MitoTEMPOL和mitovitE等化合物旨在通过在线粒体内源性途径减轻氧化应激。然而,其穿透BBB的能力有限仍是主要障碍(Hara, 2019)。此外,抗氧化治疗需谨慎调控,因过量抗氧化活性可能扰乱维持正常细胞功能所需的必需氧化信号通路(Hara, 2019)。
线粒体分裂与融合的动态过程对维持线粒体健康至关重要。这些过程的失调已被证实与多种神经退行性疾病相关。针对参与线粒体动态的蛋白质(如Drp1或OPA1)进行治疗干预正逐渐受到关注。例如,小分子Mdivi-1通过抑制Drp1,可在早期阿尔茨海默病小鼠模型中恢复线粒体形态、改善动态、减少淀粉样病理并提升认知功能,这可能通过减少Aβ生成实现(Wang, 2020)。
另一种新兴治疗策略聚焦于调节线粒体生物能代谢。增强线粒体功能和ATP生成可能有助于补偿神经退行性疾病中常见的能量缺陷。琥珀酸(OAA),作为三羧酸循环的中间产物,已被证实能增加线粒体生物能代谢流并改善脑功能。尽管在AD患者的I期临床试验中证实了其安全性,但认知益处有限(Swerdlow, 2016)。需进一步研究以确定有效剂量和长期疗效。另一候选药物CP2是一种选择性积累于神经元线粒体并抑制线粒体复合物I的化合物,在AD小鼠模型中通过减少淀粉样斑块及tau病理并维持认知功能显示出潜力(Zhang, 2015)。
基于肽的疗法,如SS-31(也称为Elamipretide或MTP-131),代表了另一种创新方法。SS-31靶向线粒体内膜并结合对线粒体膜完整性至关重要的脂质——心磷脂。这种结合支持参与氧化磷酸化(OXPHOS)的超复合体的形成。前临床研究表明,SS-31可改善AD模型中的线粒体和突触功能,并已近期在罕见疾病(如弗里德赖希共济失调症)的临床试验中进行测试(NCT05168774)。
综上所述,针对线粒体功能障碍的治疗策略在治疗神经退行性疾病方面展现出巨大潜力。然而,仍面临诸多挑战,包括优化药物递送、确保血脑屏障穿透性,以及在抑制活性氧(ROS)与维持生理信号传导之间取得平衡。持续研究对于完善这些疗法并将其转化为治疗AD、PD和ALS等疾病的有效治疗手段至关重要。
我们的团队很乐意解答关于小胶质细胞和星形胶质细胞线粒体功能障碍及神经退行性疾病的任何问题,或提供我们用于治疗效果研究的阿尔茨海默病(AD)、肌萎缩侧索硬化症(ALS)和帕金森病(PD)动物模型相关具体信息。
相关内容
关于小胶质细胞和星形胶质细胞线粒体功能障碍与神经退行性疾病的最新研究进展,以及在神经退行性疾病动物模型中评估治疗药物时的最佳实践。
小胶质细胞与神经元相互作用与神经退行性疾病
对小胶质细胞与神经元之间直接相互作用的简要综述,以及这些细胞间相互作用在神经退行性疾病中可能受到的影响。
小胶质细胞、星形胶质细胞与α-突触核蛋白在帕金森病中的作用
α-synuclein在帕金森病及其他α-synuclein相关疾病中对小胶质细胞和星形胶质细胞的影响。
微胶质细胞和星形胶质细胞的溶酶体功能障碍
关于小胶质细胞和星形胶质细胞溶酶体功能障碍及其在神经退行性疾病中作用的概述。
神经退行性疾病中的TNF-α和微胶质细胞
概述肿瘤坏死因子-α(TNF-α)在小胶质细胞中的作用及其对神经退行性病变进展的影响。
线粒体功能障碍与帕金森病
概述线粒体功能障碍与帕金森氏病神经退行性病变的关系。
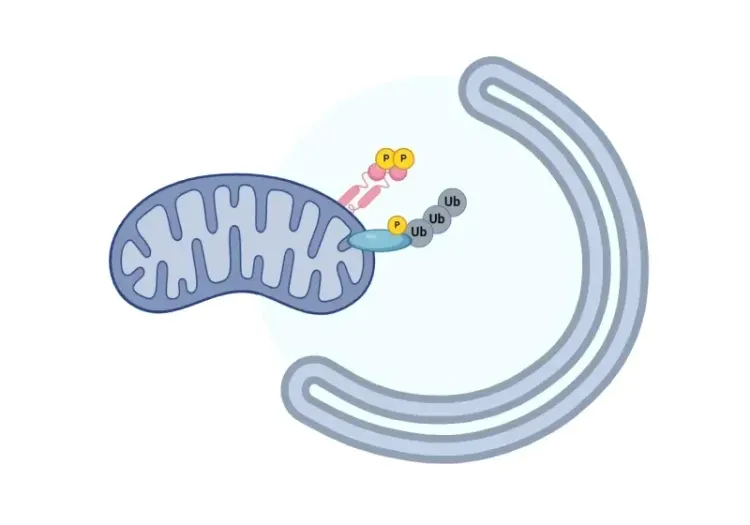
线粒体吞噬与帕金森病
概述受损的线粒体吞噬如何导致帕金森病的神经变性。