신경퇴행성 질환에서의 미세아교세포의 자식작용 장애
미세아교세포의 자식작용 장애가 신경퇴행성 질환의 진행에 어떻게 기여하는가.
미세아교세포의 오토파지는 무엇인가?
미세아교세포는 중추신경계(CNS)의 상주 면역 세포로, 뇌의 항상성 유지에 필수적입니다. 이 고도로 동적인 세포들은 혈관-뇌 장벽(BBB) 조절, 시냅스 정제, 면역 반응 조정에 기여합니다. 환경을 지속적으로 감시하는 미세아교세포는 병원체, 세포 잔여물, 단백질 집합체를 수용체(예: 톨 유사 수용체(TLRs))를 통해 탐지하고 제거합니다.
신경독성 단백질 집합체가 축적되면, 이들은 인터루킨-1β(IL-1β)와 종양 괴사 인자-α(TNF-α)와 같은 염증성 사이토카인의 분비를 자극합니다. 이 상황은 염증성 환경을 조성하여 단백질 변형과 응집을 가속화하며, 신경염증과 신경세포 손상의 악순환을 촉진합니다. 대부분의 연구는 신경세포에 초점을 맞췄지만, 단백질 응집체는 미세아교세포를 포함한 글리아 세포에서도 축적되어 신경퇴행성 병리학에 관여합니다.
자식작용과 식작용은 두 가지 주요 세포 분해 과정입니다. 포식작용은 주로 세포 외 잔여물을 제거하는 반면, 오토파지는 손상된 세포 소기관과 변형된 단백질 등 세포 내 구성 요소를 표적으로 삼아 리소좀으로 이동시켜 분해합니다. 미세아교세포에서 오토파지는 세포 내 품질 관리에 기여할 뿐만 아니라 면역 활동 조절에도 관여합니다. 이 기능은 노화 과정에서 오토파지 효율이 감소함에 따라 점점 더 중요해지며, 이는 노화의 대표적인 특징으로 알려져 있습니다.
자식작용은 세 가지 주요 형태로 구분됩니다(Jülg, 2021). 미세자식작용은 리소좀에 의해 세포질 내 물질이 직접 삼키는 과정입니다. 분자 샤페론에 의해 조절되는 자식작용은 샤페론 인식에 따라 특정 단백질을 선택적으로 표적화합니다. 가장 널리 연구된 형태인 거대자식작용은 이중막을 가진 자식소체를 형성하여 세포 성분을 삼킨 후 리소좀과 융합하여 자식소체를 형성해 분해합니다.
대식작용은 자식작용 관련(ATG) 단백질에 의해 엄격히 조절됩니다. 이 과정에는 ULK1 복합체의 활성화, Beclin1-PIK3C3 복합체의 형성, LC3-I에서 LC3-II로의 전환이 포함됩니다(Jülg, 2021). 주요 조절 인자로는 오토파지를 억제하는 포유류 라파마이신 표적 복합체 1(mTORC1)과 이를 촉진하는 AMP 활성화 단백질 키나제(AMPK)가 있습니다. 적절한 오토파지는 이러한 반대 신호 간의 균형을 유지하는 데 달려 있습니다(Lin, 2023; Ou-Yang, 2023). 미세아교세포에서는 추가 조절 경로로 p38 MAPK 신호전달 경로와 오토파지 장애의 지표로 자주 사용되는 적응 단백질 p62가 포함됩니다(Lin, 2023; Ou-Yang, 2023).
최근 연구는 미세아교세포의 오토파지가 중추신경계(CNS) 염증을 조절하는 데 중요한 역할을 한다는 점을 강조합니다(Zhu, 2022). 오토파지가 손상되면 미세아교세포의 필수 기능인 식작용과 면역 조절이 저하되어 만성 신경염증이 발생합니다(Zhu, 2022). 신경염증은 많은 신경퇴행성 질환의 특징적인 특징이기 때문에, 자식작용은 염증 매개체가 유해 수준에 도달하기 전에 제거함으로써 보호 메커니즘으로 작용합니다. 이 시스템이 실패하면 지속적인 염증과 신경세포 손상이 발생합니다(Lin, 2023).
미세아교세포 오토파지의 주요 항염증 기능 중 하나는 염증성 사이토카인을 활성화하는 다중 단백질 복합체인 NLRP3 염증체( inflammasome)의 억제입니다. 오토파지 장애는 NLRP3 활성화 증가를 초래해 신경독성에 기여합니다. 이 관계는 상호작용적입니다—NLRP3 활성은 오토파지를 방해해 CNS 면역 조절 장애에 핵심 역할을 하는 피드백 루프를 형성합니다.
신경세포 중심의 연구가 자식소체 연구를 주도해 왔지만, 미세아교세포의 자식소체 기능 장애가 신경퇴행성 질환에 크게 기여한다는 증거가 증가하고 있습니다. 미세아교세포의 자식소체 활성 증진은 치료적 접근법으로 연구되고 있습니다. 변형된 단백질의 제거를 개선하고 만성 염증을 완화함으로써, 미세아교세포의 자식소체를 회복시키는 것은 단백질 응집과 면역 불균형으로 인한 질환의 진행을 늦추거나 예방할 수 있습니다.
신경퇴행성 질환(예: 알츠하이머 병, 파킨슨 병, 루게릭 병)에서 미세아교세포의 오토파지가 어떻게 손상되는가?
알츠하이머 병(AD)
AD는 가장 흔한 신경퇴행성 질환으로, 진행성 인지 기능 저하와 세포외 아밀로이드-베타(Aβ) 플라크 및 세포내 타우 엉킴의 축적이 특징입니다. 미세아교세포는 AD에서 병리적 집합체의 제거에 참여하는 동시에 염증에 기여하는 이중 역할을 합니다(Cho, 2014).
AD에서 미세아교세포 기능과 관련된 주요 오토파지 관련 조절인자에는 PPAR-α, Atg1, Map1lc3b, 및 BECN1이 포함됩니다. PPAR-α의 활성화는 인간 미세아교세포 및 AD 마우스 모델에서 오토파지를 증가시켜 Aβ 플라크 부하 감소, 인지 기능 개선, 및 플라크 주변 글리아 활동 증가를 유발합니다(Luo, 2020). 반면, Atg1 또는 Map1lc3b를 억제하면 실험 모델에서 Aβ 분해가 저해되고 염증 신호전달이 강화됩니다(Cho, 2014)
마우스에서 BECN1 기능의 부분적 상실은 미세아교세포의 NLRP3 염증체 활성 증가와 IL-1β 및 IL-18 생산 증가를 유발합니다(Houtman, 2019). APP/PS1 마우스에서 BECN1 발현이 감소하면 NLRP3 및 카스파제-1 활성화가 증가합니다. 초고해상도 이미징 연구는 NLRP3와 LC3 양성 소체 간의 공위치를 확인했으며, 이는 자식작용이 염증체 구성 요소를 직접 분해할 수 있음을 시사합니다(Houtman, 2019).
이러한 결과는 인간 데이터로 뒷받침됩니다. 알츠하이머 병 환자로부터 분리된 미세아교세포는 Beclin1 수준이 감소되어 있으며, 이는 자포소체 형성 및 식작용 기능 장애로 인해 Aβ 제거 장애에 기여할 가능성이 있습니다 (Lucin, 2013). Aβ 식작용 후 미세아교세포는 LC3-II와 적응 단백질 옵티뉴린(OPTN)을 포함한 메커니즘을 통해 이를 분해하며, 이는 STK11/LKB1-AMPKα 경로에 의해 조절됩니다. 이러한 통찰은 AD에서 Aβ 축적과 염증을 제한하는 데 있어 정상적인 자포작용의 필수적 역할을 강화합니다.
파킨슨병(PD)
PD는 흑질 밀집부(SNc)의 도파민 신경세포의 진행성 퇴화로 특징지어지는 두 번째로 흔한 신경퇴행성 질환입니다. 임상적으로 PD는 떨림, 경직, 운동 느림, 자세 불안정과 같은 운동 증상 및 인지 기능 저하, 수면 장애와 같은 비운동 증상을 동반합니다. 병리학적 특징은 Lewy 체와 신경돌기 내 α-synuclein의 축적입니다.
미세아교세포의 자식작용은 염증 조절과 α-synuclein 제거에 핵심적 역할을 합니다. 과도한 α-synuclein은 자식작용 흐름을 방해하고 산화 스트레스를 유발합니다. 가족성 PD와 연관된 DJ-1 유전자는 미세아교세포의 자식작용을 조절합니다. 이 유전자의 결손은 α-synuclein 제거를 방해하고 염증을 강화합니다(Nash, 2017). 미세아교세포에서 Atg5를 삭제하면 MPTP 모델에서 NLRP3 활성화가 증가하고 신경세포 손실이 악화됩니다(Qin, 2021). Drp1을 약리학적으로 억제하면 자식작용이 회복되고 α-synuclein 축적이 감소합니다(Fan, 2019).
세포외 알파-시누클린은 TLR4 매개 p38 및 Akt/mTOR 신호전달 경로를 통해 자식작용을 억제합니다. 이 중 어느 경로를 차단해도 자식작용 기능을 회복시킵니다(Tu, 2021). 미세아교세포는 TLR4 신호전달과 NF-κB에 의해 유도된 p62 발현에 의존하는 선택적 자식작용 과정인 'synucleinphagy'를 통해 신경세포에서 방출된 α-synuclein을 분해합니다 (Choi, 2020).
노화는 이러한 장애를 악화시킵니다. 노화된 쥐는 인간 α-synuclein 주사 후 젊은 쥐보다 α-synuclein을 더 오래 유지하며, 이는 질병 진행에 기여하는 연령 관련 자식작용 효율 저하를 시사합니다(Hong, 2024).
근위축성 측삭경화증(ALS)
ALS는 운동 신경 세포의 손실로 특징지어지는 진행성 및 치명적인 신경퇴행성 질환입니다. 세포질 내 TDP-43 침착물을 특징으로 하며, C9ORF72 및 SOD1 유전자 변이와 유전적으로 연관되어 있습니다. 미세아교세포의 활성화 및 기능 장애는 ALS 병리학에서 두드러지지만, 자식작용의 구체적인 역할은 덜 이해되고 있습니다.
인간 유도 만능 줄기세포(hiPSC)에서 유래한 PFN1 돌연변이 미세아교세포 유사 세포(hiPSC-MG)는 자식작용과 식작용이 손상되어 있습니다(Funes, 2024). 이러한 결함은 라파마이신에 의해 역전되며, 이는 오토파지 의존적 메커니즘을 시사합니다(Funes, 2024). C9ORF72 돌연변이를 가진 hiPSC 유래 미세아교세포는 오토파지 감소와 면역 활동 증가를 보이며, 약리학적 오토파지 자극은 운동 신경 세포 생존을 개선합니다(Banerjee, 2023).
SOD1G93A ALS 마우스와 LC3 리포터 계통을 교배한 모델에서 증상 발현 단계에서 미세아교세포와 아스트로사이트의 유동성이 증가하지만, 올리고덴드로사이트에서는 이 반응이 관찰되지 않습니다(Perera, 2025). 또한, 자식작용 장애는 운동 피질보다 척수에서 더 일찍 나타납니다. 이는 질병 진행에 있어 지역적 및 세포 유형 특이성을 시사합니다(Perera, 2025).
알츠하이머병(AD), 파킨슨병(PD), ALS에서 얻어진 증거는 미세아교세포의 자식작용 장애가 단백질 축적과 만성 염증을 촉진하여 신경 퇴화를 가속화하는 공통된 메커니즘을 시사합니다. 따라서 미세아교세포의 자식작용을 강화하는 것은 질병 진행을 늦추는 유망한 전략이 될 수 있습니다.
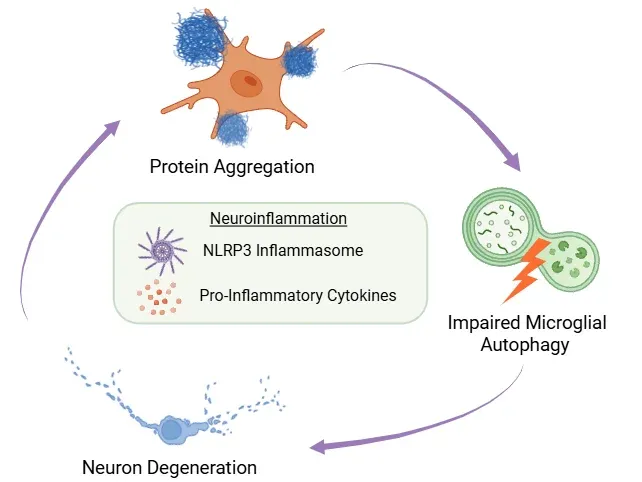
잘못 접힌 단백질의 축적, 미세아교세포의 자식작용 장애, 및 신경세포 퇴화 사이의 피드백 루프를 보여주는 그림. 이 사이클의 중심에는 NLRP3 염증체 활성화와 염증성 사이토킨 분비에 의해 유발되는 신경염증이 위치해 있습니다.
미세아교세포의 자식작용을 신경퇴행성 질환의 치료 표적으로 삼는 방법은 무엇인가?
미세아교세포의 자식작용을 표적화하는 것은 알츠하이머 병(AD), 파킨슨 병(PD), 루게릭 병(ALS)과 같은 신경퇴행성 질환의 치료를 위한 유망한 치료 전략으로 제시되고 있습니다. 현재까지 미세아교세포의 자식작용을 선택적으로 조절하는 승인된 치료법은 없지만, 전임상 연구 결과는 자식작용 활성화를 통해 병리적 단백질 제거를 개선하고 염증을 감소시키며 중추신경계(CNS)의 균형을 회복할 수 있다는 가능성을 뒷받침하고 있습니다.
여러 약리학적 제제가 연구되었습니다. mTOR 억제제인 라파마이신은 자식작용을 유도하고 TLR2 매개 α-시누클린 발현을 감소시킵니다(Dzamko, 2017). 그러나 ALS 모델에서 라파마이신은 운동 신경 세포 손실을 악화시켰으며(Zhang, 2011), 임상 시험에서는 유의미한 혜택을 입증하지 못했습니다(Mandrioli, 2023). AMPK를 활성화하는 메트포르민은 α-시누클린 응집을 감소시켜 파킨슨병 모델에서 신경 보호 효과를 보여주었으나, 미세아교세포의 자식작용에 대한 직접적인 영향은 명확하지 않습니다(Lu, 2016). 플루옥세틴은 LC3-II 수준과 자식소체 형성을 증가시켜 미세아교세포의 자식작용을 촉진하며, 염증 감소와 응집체 제거에 잠재적 효과를 보였습니다(Park, 2021). 카엠페롤은 미세아교세포의 자식작용을 촉진하고 NLRP3 활성화를 억제하여 미세아교세포의 사이토킨 분비를 감소시킵니다(Han, 2019). 자식작용 유도제인 트레할로스는 초기 ALS 연구에서 유망했으나, 최근 임상 시험 결과는 결론적이지 않았습니다(HEALEY ALS Platform Trial, 2025).
이러한 혼합된 결과에 따라 연구자들은 세포 유형 특이적 전달 전략을 개발 중입니다. 혈뇌 장벽(BBB)을 통과하고 미세아교세포를 표적하는 나노입자(예: MCPZFS NPs)는 Aβ 제거를 촉진하고 사이토킨 분비를 감소시키는 능력을 보여주었습니다. 이러한 입자는 단백질 분해를 자식작용에서 프로테아좀 경로로 재지향시킬 수 있습니다(Liu, 2019). 그러나 BBB 침투, 표적 정확도, 나노입자 변이성 등 도전 과제가 남아 있습니다(Lin, 2023).
탄소 기반 나노물질인 그래핀 산화물(GO)은 AMPK 활성화를 통해 mTOR 신호전달을 억제함으로써 오토파지를 촉진합니다. GO는 질병 모델에서 미세아교세포의 Aβ 제거를 향상시키고 신경독성을 감소시키는 것으로 나타났습니다(Li, 2020).
유전자 치료 접근법도 탐구 중입니다. 글리아 세포 특이적 프로모터를 갖춘 아데노연관 바이러스(AAV) 벡터는 미세아교세포에서 자식작용 유전자의 표적 조절을 가능하게 합니다(O’Carroll, 2021). 이 접근법은 특히 BBB를 통과하는 어려움 고려 시 전신 약물보다 장점을 제공합니다.
자율적 소화를 유도하는 약물과 염증체 억제제를 결합하면 시너지 효과를 기대할 수 있습니다. 두 경로를 동시에 표적화하는 접근법은 단독 치료보다 단백질 축적과 염증을 더 효과적으로 억제하는 것으로 나타났습니다(Wang, 2023).
요약하면, 미세아교세포의 자식작용을 조절하는 대부분의 전략은 여전히 초기 개발 단계에 있지만, 이 분야는 빠르게 발전하고 있습니다. 전달 시스템의 정교화, 세포 특이성 강화, 장기적 안전성 확립을 위한 추가 연구가 필요합니다. 미세아교세포의 자식작용을 회복시키는 치료법은 단백질 병리와 면역 이상을 동시에 해결함으로써 신경퇴행을 늦추는 데 큰 잠재력을 가지고 있습니다.
저희 팀은 신경퇴행성 질환에서 미세아교세포의 자식작용 장애에 대한 질문에 답변해 드리거나, 치료 효과 연구에 사용되는 모델에 대한 구체적인 정보를 제공해 드릴 수 있습니다.
신경퇴행성 질환 모델에 대해 더 알아보세요
관련 콘텐츠
신경염증에 대한 최신 정보 및 신경퇴행성 질환 동물 모델에서 치료제 평가와 관련된 최선의 실천 방법.
미세아교세포와 성상교세포의 리소좀 기능 장애
미세아교세포와 성상교세포의 리소좀 기능 장애와 신경퇴행성 질환에서의 그 역할에 대한 개요.
자가포식, 파킨슨병, 도파민성 신경세포
파킨슨병에서 손상된 자가포식 작용이 어떻게 도파민성 뉴런의 병리학적 변화와 신경 퇴화로 이어질 수 있는지에 대한 개요.
자가포식 및 신경퇴행성 질환
세포 자가포식이 뇌 건강과 신경 퇴화에 어떤 역할을 하는지에 대한 개요.
NLRP3 인플라마좀과 신경퇴행성 질환
NLRP3 인플라마솜과 알츠하이머병, 파킨슨병, ALS를 포함한 신경 퇴행성 질환에서의 그 역할에 대한 개요.
신경 퇴행성 질환의 TNF-α & 미세 아교 세포
미세아교세포에서 종양괴사인자-알파(TNF-α)의 기능과 신경퇴행의 진행에 대한 기여에 대한 개요.
자가포식 및 전사 인자 EB(TFEB)
전사 인자 EB(TFEB)와 자가포식 및 신경 퇴행성 질환에서의 역할에 대한 개요.