ミクログリアオートファジーとは何ですか?
ミクログリアは中枢神経系(CNS)の常在免疫細胞であり、脳の恒常性維持に不可欠です。これらの高度に動的な細胞は、血液脳関門(BBB)の調節、シナプスの剪定、免疫応答の調整などに寄与しています。環境を継続的に監視することで、ミクログリアは病原体、細胞残渣、タンパク質凝集体を認識し、トール様受容体(TLR)などの受容体を介して排除します。
神経毒性タンパク質凝集体が蓄積すると、インターロイキン-1β(IL-1β)や腫瘍壊死因子-α(TNF-α)などのプロ炎症性サイトカインの放出を刺激します。この状況は、さらにタンパク質の異常折り畳みと凝集を加速する炎症促進環境を生み出し、神経炎症と神経細胞損傷の悪循環を助長します。これまでの研究は主に神経細胞に焦点を当ててきましたが、タンパク質凝集体はミクログリアを含むグリア細胞にも蓄積し、神経変性病理に関与していることが示唆されています。
オートファジーとファゴサイトーシスは、細胞の分解プロセスにおける2つの主要なプロセスです。ファゴサイトーシスは主に細胞外デブリを除去する一方、オートファジーは損傷したオルガネラや異常なタンパク質などの細胞内成分を標的とし、それらをリソソームに輸送して分解します。ミクログリアでは、オートファジーは細胞内品質管理に貢献するだけでなく、免疫活動の調節にも関与しています。この機能は加齢に伴い重要性が増します。なぜなら、オートファジーの効率は加齢とともに低下するからです。これは加齢の代表的な特徴の一つです。
オートファジーには3つの主要な形態があります(Jülg, 2021)。ミクロオートファジーは、リソソームによる細胞質物質の直接的な取り込みです。シャペロン介在オートファジーは、シャペロン認識に基づいて特定のタンパク質を選択的に標的とします。最も広く研究されているタイプであるマクロオートファジーは、細胞成分を取り込む二重膜のオートファゴソームの形成を伴い、その後リソソームと融合してオートリソソームを形成し、分解されます。
マクロオートファジーは、オートファジー関連(ATG)タンパク質によって厳密に調節されています。このプロセスには、ULK1複合体の活性化、Beclin1-PIK3C3複合体の形成、およびLC3-IからLC3-IIへの変換が含まれます(Jülg, 2021)。主要な調節因子には、オートファジーを抑制する哺乳類ラパマイシン標的複合体1(mTORC1)と、それを促進するAMP活性化プロテインキナーゼ(AMPK)が含まれます。適切なオートファジーは、これらの対立するシグナルのバランスを維持することに依存しています(Lin, 2023;Ou-Yang, 2023)。ミクログリアでは、追加の調節経路としてp38 MAPKシグナル伝達とアダプタータンパク質p62が含まれ、これらはオートファジー障害のマーカーとしてよく使用されます(Lin, 2023;Ou-Yang, 2023)。
最近の研究は、ミクログリアのオートファジーが中枢神経系(CNS)炎症の調節に重要な役割を果たすことを示しています(Zhu, 2022)。オートファジーが障害を受けると、ミクログリアの重要な機能である貪食作用や免疫調節が障害され、慢性神経炎症を引き起こします(Zhu, 2022)。神経炎症は多くの神経変性疾患の定義的な特徴であるため、オートファジーは炎症性メディエーターが有害なレベルに達する前に除去する保護メカニズムとして機能します。このシステムが機能不全に陥ると、持続的な炎症と神経細胞損傷が引き起こされます(Lin, 2023)。
ミクログリアオートファジーの主要な抗炎症機能の一つは、プロ炎症性サイトカインを活性化させる多タンパク質複合体であるNLRP3炎症小体(NLRP3 inflammasome)の抑制です。オートファジーの障害はNLRP3の活性化を促進し、神経毒性に寄与します。この関係は相互作用的であり、NLRP3の活性もオートファジーを障害し、中枢神経系(CNS)の免疫調節障害に重要な役割を果たすフィードバックループを形成します。
オートファジー研究は神経細胞に焦点を当てた研究が主流でしたが、ミクログリアのオートファジー機能障害が神経変性疾患に大きく関与するとの見方が、増加するデータによって支持されています。ミクログリアのオートファジー活性を高めることは、現在治療アプローチとして検討されています。異常なタンパク質の除去を改善し、慢性炎症を軽減することで、ミクログリアのオートファジーを回復させることは、タンパク質凝集と免疫バランスの乱れの両方によって引き起こされる疾患の進行を遅らせたり、予防したりする可能性があります。
神経変性疾患(AD、PD、ALSなど)において、ミクログリアのオートファジーはどのように障害されるのでしょうか?
アルツハイマー病(AD)
ADは最も一般的な神経変性疾患であり、進行性の認知機能低下と、細胞外のアミロイドβ(Aβ)プラークおよび細胞内のタウタンパク質の凝集物の蓄積が特徴です。ミクログリアはADにおいて二重の役割を果たしており、病理学的凝集物の除去に関与する一方、炎症にも寄与しています(Cho, 2014)。
ADにおけるミクログリアの機能に関与する主要なオートファジー関連調節因子には、PPAR-α、Atg1、 Map1lc3b、および BECN1が含まれます。PPAR-αの活性化は、ヒトのミクログリアおよびADマウスモデルにおいてオートファジーを促進し、Aβプラークの蓄積を減少させ、認知機能の改善、およびプラーク周辺のグリア細胞の活性を高めます(Luo, 2020)。一方、Atg1または Map1lc3bのノックダウンは、実験モデルにおいてAβの分解を阻害し、炎症シグナル伝達を強化します(Cho, 2014)
マウスにおけるBECN1機能の部分的な喪失は、ミクログリアのNLRP3炎症小体活性の上昇とIL-1βおよびIL-18の産生増加を引き起こします(Houtman, 2019)。BECN1発現が 低下したAPP/PS1マウスでは、NLRP3とカスパーゼ-1の活性化がより顕著に観察されます。超解像イメージング研究は、NLRP3とLC3陽性小胞の共局在を確認し、オートファジーが炎症小体成分を直接分解する可能性を示唆しています(Houtman, 2019)。
これらの結果はヒトのデータによって支持されています。アルツハイマー病患者から分離されたミクログリアではBeclin1のレベルが低下しており、これはオートファゴソームの形成と貪食機能の障害によりAβのクリアランスが障害されることに寄与していると考えられます(Lucin, 2013)。Aβの食作用後、ミクログニアはLC3-IIとアダプタータンパク質オプティヌリン(OPTN)を介したメカニズムによりAβを分解し、このプロセスはSTK11/LKB1-AMPKα経路によって調節されています。これらの知見は、ADにおけるAβの蓄積と炎症を抑制する上で、健全なオートファジーの不可欠な役割を強化しています。
パーキンソン病(PD)
PDは、黒質緻密部(SNc)のドーパミン神経細胞の進行性変性を特徴とする、2番目に多い神経変性疾患です。臨床的には、振戦、筋強剛、動作緩慢、姿勢不安定などの運動症状に加え、認知機能低下や睡眠障害などの非運動症状が現れます。病理学的特徴は、Lewy体と神経突起にα-シヌクレインが蓄積することです。
ミクログリアのオートファジーは、炎症の調節とα-シヌクレインの除去に不可欠です。過剰なα-シヌクレインはオートファジーの流れを妨げ、酸化ストレスを引き起こします。家族性パーキンソン病と関連するDJ-1遺伝子は、ミクログリアのオートファジーを調節します。その欠失はα-シヌクレインの除去を妨げ、炎症を悪化させます(Nash, 2017)。ミクログリアにおけるAtg5の欠失は、MPTPモデルにおいてNLRP3の活性化を増加させ、神経細胞の喪失を悪化させます(Qin, 2021)。Drp1を薬理学的に阻害すると、オートファジーが回復し、α-シヌクレインの蓄積が減少します(Fan, 2019)。
細胞外α-シヌクレインは、TLR4を介したp38およびAkt/mTORシグナル伝達経路を介してオートファジーを阻害します。これらの経路のいずれかを阻害すると、オートファジー機能が回復します(Tu, 2021)。ミクログリアは、TLR4シグナル伝達とNF-κB依存性p62誘導に依存する、神経細胞から放出されたα-シヌクレインを分解する選択的オートファジープロセスである「シヌクレインファジー」にも関与します(Choi, 2020)。
加齢はこれらの障害を悪化させます。ヒトα-シヌクレインを投与したマウスでは、高齢マウスは若年マウスよりもα-シヌクレインを長く保持し、加齢に伴うオートファジー効率の低下が疾患進行に寄与することを示唆しています(Hong, 2024)。
筋萎縮性側索硬化症(ALS)
ALSは、運動神経細胞の喪失を特徴とする進行性で致死性の神経変性疾患です。TDP-43の細胞質内包涵体を特徴とし、C9ORF72とSOD1の遺伝子変異と遺伝的に関連しています。ALSの病理においてミクログリアの活性化と機能障害は顕著ですが、オートファジーの具体的な役割は十分に理解されていません。
PFN1変異を有するヒト誘導多能性幹細胞(hiPSC)由来のミクログリア様細胞(hiPSC-MG)は、オートファジーとファゴサイトーシスの障害を示します(Funes, 2024)。これらの欠損はラパマイシンで逆転し、オートファジー依存性メカニズムを示唆しています(Funes, 2024)。C9ORF72変異を有するhiPSC由来ミクログリアではオートファジーが低下し免疫活性が増加しますが、薬理学的刺激によるオートファジーの活性化は運動神経細胞の生存を改善します(Banerjee, 2023)。
SOD1G93AALSマウスとLC3レポーターラインを交配したモデルでは、症状発現期にミクログリアとアストロサイトの流動が増加しますが、オリゴデンドロサイトではこの反応は認められません(Perera, 2025)。さらに、オートファジーの機能障害は運動皮質よりも脊髄で早期に発現し、疾患進行における地域的および細胞タイプ特異性を示唆しています(Perera, 2025)。
AD、PD、ALSからの証拠は、ミクログリアのオートファジー障害がタンパク質蓄積と慢性炎症を促進し、神経変性を加速する共通のメカニズムを示唆しています。ミクログリアのオートファジーを強化することは、したがって、疾患進行を遅らせるための有効な戦略となる可能性があります。
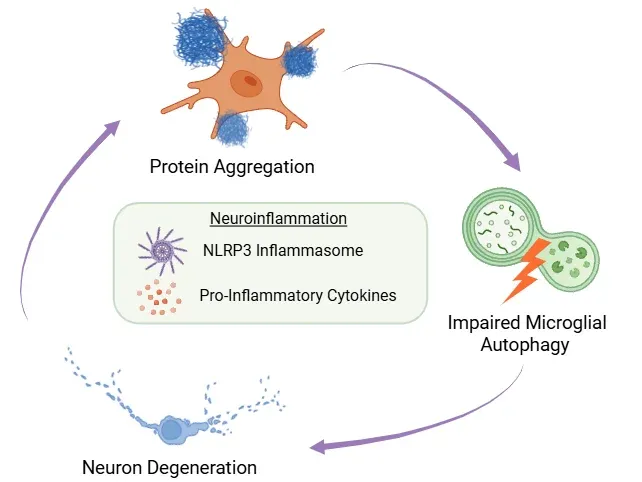
誤って折りたたまれたタンパク質の蓄積、ミクログリアのアポトーシスの障害、および神経細胞の変性の間のフィードバックループの図解です。このサイクルの中心には、NLRP3 インフラマソームの活性化と炎症誘発性サイトカインの放出によって引き起こされる神経炎症があります。
神経変性疾患の治療において、ミクログリアのアポトーシスを標的とする方法はどのようなものがあるでしょうか?
ミクログリアのアポトーシスを標的とするアプローチは、アルツハイマー病(AD)、パーキンソン病(PD)、筋萎縮性側索硬化症(ALS)などの神経変性疾患の治療において有望な治療戦略です。現在、ミクログリアのアポトーシスを選択的に調節する承認された治療法はありませんが、前臨床研究の証拠は、アポトーシス活性を高めることが病理学的タンパク質の除去を促進し、炎症を軽減し、中枢神経系(CNS)の恒常性を維持する可能性を示唆しています。
いくつかの薬理学的薬剤が検討されています。mTOR阻害剤であるラパマイシンはオートファジーを誘導し、TLR2介在性α-シヌクレインの発現を減少させます(Dzamko, 2017)。しかし、ALSモデルではラパマイシンが運動神経細胞の喪失を悪化させ(Zhang, 2011)、臨床試験では有意な効果は示されていません(Mandrioli, 2023)。AMPKを活性化させるメトホルミンは、PDモデルにおいてα-シヌクレインの凝集を減少させることで神経保護効果を示しましたが、ミクログリアのオートファジーへの直接的な影響は不明です(Lu, 2016)。フルオキセチンは、LC3-IIレベルとオートファゴソームの形成を増加させることでミクログリアのオートファジーを促進し、炎症の軽減と凝集物の除去に潜在的な効果があります(Park, 2021)。ケンフェロールは、ミクログリアのオートファジーを促進し、NLRP3 の活性化を抑制することで、ミクログリアにおけるサイトカインの分泌を減少させます(Han、2019)。オートファジー誘導剤であるトレハロースは、初期の ALS 研究では有望でしたが、最近の臨床試験の結果は決定的なものとはなりませんでした(HEALEY ALS Platform Trial、2025)。
こうしたさまざまな結果を受けて、研究者たちは細胞タイプに特化した送達戦略の開発を進めています。MCPZFS NP など、血液脳関門を通過してミクログニアを標的とするナノ粒子は、Aβのクリアランスを高め、サイトカインの放出を抑制する能力があることが示されています。これらの粒子は、タンパク質の分解をオートファジーからプロテアソーム経路にリダイレクトする可能性があります(Liu、2019)。しかし、BBB 透過性、標的の精度、ナノ粒子の変動性などの課題が残っています(Lin、2023)。
炭素系ナノ材料であるグラフェン酸化物(GO)は、AMPK活性化を介してmTORシグナル伝達を阻害することでオートファジーを促進します。GOは、疾患モデルにおいてミクログリアのAβクリアランスを促進し、神経毒性を軽減することが示されています(Li, 2020)。
遺伝子療法アプローチも探索されています。グリア細胞特異的プロモーターを有するアデノ随伴ウイルス(AAV)ベクターは、ミクログリアにおけるオートファジー遺伝子の標的調節を可能にします(O’Carroll, 2021)。このアプローチは、特にBBB透過の困難性を考慮すると、全身投与薬に比べて優位性があります。
オートファジー誘導剤と炎症小体阻害剤を組み合わせることで、相乗効果が期待できます。両経路を同時に標的とするアプローチは、いずれかの介入単独に比べて、タンパク質蓄積と炎症の抑制がより効果的であることが示されています(Wang, 2023)。
要約すると、ミクログリアのオートファジーを調節するほとんどの戦略は依然として初期開発段階にあるものの、この分野は急速に進展しています。デリバリーシステムの精緻化、細胞特異性の向上、長期的な安全性の確立に向けた継続的な研究が求められています。ミクログリアのオートファジーを回復させる療法は、タンパク質病変と免疫異常の両方を標的とすることで、神経変性の進行を遅らせる大きな可能性を秘めています。
当社のチームは、神経変性疾患におけるミクログリアのアポトーシスの障害に関するご質問、または治療効果の研究に使用しているモデルに関する具体的な情報について、喜んでお答えいたします。
当社の神経変性疾患モデルについて詳しく見る
関連コンテンツ
神経炎症に関する最新情報と、神経変性疾患の動物モデルにおける治療薬の評価に関するベストプラクティス。
ミクログリアとアストロサイトのライソゾーム機能不全
ミクログリアとアストロサイトのライソゾーム機能不全の概要と、神経変性疾患におけるその役割。
オートファジー、パーキンソン病、ドーパミン作動性ニューロン
パーキンソン病における障害のあるオートファジーが、ドーパミン作動性ニューロンにおける病理学的変化と神経変性につながる仕組みの概要。
オートファジーと神経変性疾患
細胞性オートファジーが脳の健康と神経変性において果たす役割についての概要。
NLRP3インフラマソームと神経変性疾患
NLRP3インフラマソームの概要と、アルツハイマー病、パーキンソン病、ALSなどの神経変性疾患におけるその役割について。
神経変性疾患におけるTNF-αとミクログリア
ミクログリアにおける腫瘍壊死因子-α(TNF-α)の機能と、神経変性進行への寄与についての概要。
オートファジーと転写因子EB(TFEB)
転写因子TFEB(Transcription Factor EB)の概要と、オートファジーおよび神経変性疾患における役割について。