Mitophagie et maladie de Parkinson
Une vue d'ensemble de la façon dont une mitophagie déficiente peut conduire à la neurodégénérescence dans la maladie de Parkinson.
Cette ressource décrit:
Qu'est-ce que la mitophagie?
L'autophagie est un processus de dégradation lysosomale qui décompose les débris cellulaires, y compris les protéines mal repliées et les organites endommagés, et recycle leurs composants pour la synthèse de nouvelles molécules (Gómez-Virgilio, 2022). La mitophagie, également connue sous le nom d'autophagie mitochondriale, est une forme spécialisée d'autophagie qui dégrade sélectivement les mitochondries endommagées et dysfonctionnelles afin de maintenir l'homéostasie énergétique (Uoselis, 2023; Wang, 2023).
Dans les mitochondries dysfonctionnelles ou endommagées, la membrane mitochondriale interne se dépolarise, ce qui entraîne l'accumulation de PINK1 (PTEN-induced kinase 1) sur la membrane externe des mitochondries endommagées (Jin, 2012; Narendra, 2012; Pickrell, 2015). PINK1 agit comme un capteur moléculaire qui recrute et active Parkin, une E3 ubiquitine ligase, qui ubiquitine plusieurs protéines de la membrane mitochondriale externe (OMM), initiant la mitophagie et marquant les mitochondries endommagées Chan, 2011; Yoshii, 2011). Ces mitochondries marquées sont ensuite encapsulées dans des vésicules autophagosomiques à double membrane, qui les transportent vers les lysosomes pour les dégrader (Youle, 2011; Jin, 2012; Pickrell, 2015).
La mitophagie joue un rôle important dans le contrôle de la qualité des mitochondries en soutenant la génération de nouvelles mitochondries saines par la biogenèse et en veillant à ce que les besoins énergétiques des cellules soient satisfaits. La fission et la fusion des mitochondries fonctionnent en tandem avec la mitophagie pour créer une population mitochondriale fonctionnelle ; la fission isole les parties endommagées des mitochondries en les fragmentant en mitochondries filles plus petites qui seront marquées pour une dégradation ciblée, tandis que la fusion optimise la fonction mitochondriale en permettant aux mitochondries de compenser leurs défauts mutuels et de soutenir la production d'énergie cellulaire (Twig, 2008; Ashrafi, 2013).
Pour surveiller et quantifier la mitophagie in vivo et in vitro, les scientifiques utilisent souvent une combinaison de techniques. Il s'agit notamment de la microscopie électronique (ME) pour la visualisation à haute résolution des autophagosomes engloutissant les mitochondries, des rapporteurs fluorescents sensibles au pH, tels que le Keima ciblant les mitochondries (mt-Keima), pour détecter les mitochondries dans les lysosomes, et du Western blotting pour mesurer les niveaux de protéines liées à la mitophagie (Zhu, 2011; Williams, 2017; Jung, 2019).
Lorsque la membrane mitochondriale interne se dépolarise, PINK1 s'accumule sur la membrane mitochondriale externe. La protéine agit comme un capteur moléculaire qui recrute et active la Parkin, initiant la mitophagie et marquant les mitochondries endommagées. Les mitochondries marquées sont encapsulées dans des vésicules d'autophagosome à double membrane, qui les transportent vers le lysosome pour être dégradées.
Quelle est la relation entre l'altération de la mitophagie et la dégénérescence des neurones dopaminergiques dans la maladie de Parkinson?
L'altération de la mitophagie est impliquée dans l'étiologie de la maladie de Parkinson. Des études génétiques ont révélé que des mutations de perte de fonction dans des protéines clés de la mitophagie - PINK1 et Parkin - induisent la pathologie des corps de Lewy et conduisent à une MP autosomique récessive à début précoce. Le gène Parkin est responsable de près de 50 % des cas familiaux de MP à début précoce (Kitada, 1998; Lücking, 2000; Valente, 2004; Gandhi, 2006; Samaranch, 2010).
PINK1 et Parkin agissent en tant que régulateurs de la fission mitochondriale, un processus essentiel pour isoler les fragments mitochondriaux endommagés qui seront ciblés par la machinerie de mitophagie (Buhlman, 2014; Ge, 2020). Les mutations de ces protéines entraînent l'accumulation de mitochondries anormalement grandes et dysfonctionnelles qui favorisent l'agrégation de l'alpha-synucléine dans les neurones dopaminergiques, une caractéristique importante de la maladie de Parkinson (Srinivasan, 2021). En outre, la perte de PINK1 et de Parkin réduit la solubilité de l'alpha-synucléine, ce qui favorise son agrégation dans les neurones dopaminergiques (Lonskaya, 2013). Simultanément, des formes mutantes de l'alpha-synucléine, telles que A53T, provoquent une défaillance de la voie de l'ubiquitine-protéasome (UPS) dans les neurones dopaminergiques, ce qui exacerbe l'agrégation de l'alpha-synucléine (McKinnon, 2020).
Le dysfonctionnement des mitochondries est également connu pour provoquer la dégénérescence sélective des neurones dopaminergiques dans la maladie de Parkinson, qui ont des besoins métaboliques particulièrement élevés en raison de leurs grands arbustes axonaux, de leur activité rythmique et du pompage du calcium qui régule la libération de dopamine (Arduíno, 2011; Rademacher, 2024). Lorsque la mitophagie est altérée dans la MP, elle conduit à l'accumulation de mitochondries structurellement et fonctionnellement dysfonctionnelles dans les neurones dopaminergiques. Ces mitochondries génèrent des niveaux élevés d'espèces réactives de l'oxygène (ROS) en raison de la production de superoxyde due à la fuite d'électrons, ont une production d'ATP réduite ou faible, provoquent une surcharge calcique et activent les voies de l'apoptose, ce qui contribue à la dégénérescence et à la perte de neurones dopaminergiques dans le mésencéphale (Liu, 2018; Zampese, 2020).
Les analyses post-mortem de tissus cérébraux de patients atteints de la maladie de Parkinson confirment le rôle des défaillances mitophagiques dans la pathologie de la maladie de Parkinson. Ces défaillances comprennent un dysfonctionnement du complexe I dans la substantia nigra pars compacta, une accumulation accrue d'ADN mitochondrial (ADNmt) en fonction de l'âge, une expression dérégulée des protéines mitochondriales et une augmentation des dommages oxydatifs causés à ces protéines (Mizuno, 1989; Bender, 2006; Grünewald, 2016; Giannoccaro, 2017). Ces résultats mettent en évidence le rôle de la mitophagie dans la pathogenèse de la MP et soulignent son potentiel en tant que cible thérapeutique pour la neurodégénérescence liée à la MP.
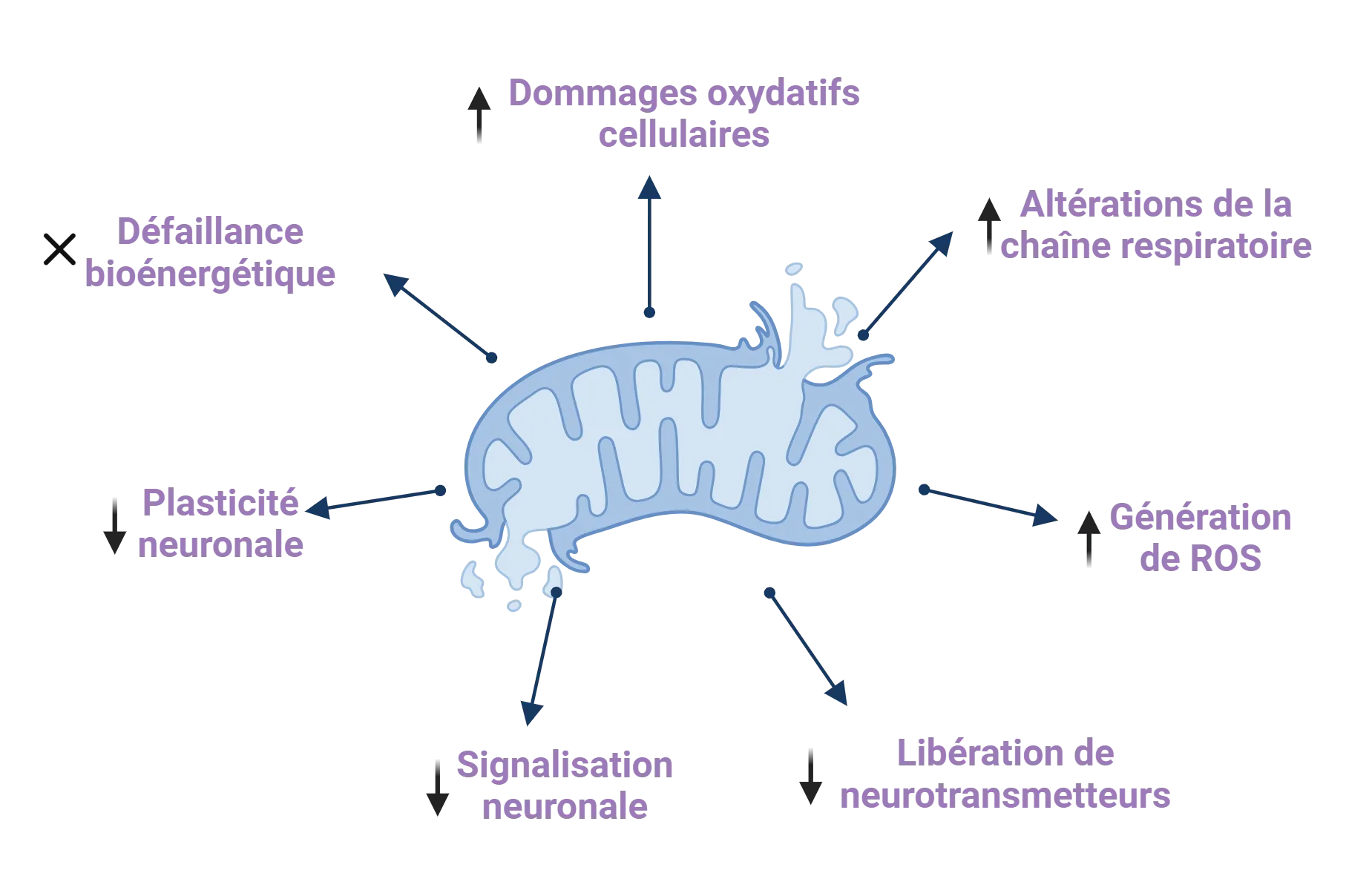
Le dysfonctionnement mitochondrial contribue au développement et à la progression de la maladie de Parkinson par une série de mécanismes. Les flèches vers le haut de l'infographie représentent les augmentations, tandis que les flèches vers le bas représentent les réductions. La figure et la légende sont reproduites de Millichap et al. (Millichap, 2021) sous la licence Creative Commons Attribution.
Les thérapies ciblant la mitophagie ont-elles montré une efficacité dans les modèles de la maladie de Parkinson?
Les défaillances de la mitophagie médiée par PINK1/Parkin ont été liées à la dégénérescence sélective et à la mort des neurones dopaminergiques dans la maladie de Parkinson (Ge, 2020). Par conséquent, la régulation pharmacologique de la mitophagie médiée par PINK1/Parkin est apparue comme une stratégie intéressante pour développer des traitements de fond visant à atténuer la perte de neurones dopaminergiques dans la maladie de Parkinson.
Des études récentes menées sur des modèles murins de la maladie de Parkinson mettent en évidence le potentiel thérapeutique des activateurs de la mitophagie PINK1/Parkin. Par exemple, dans leurs derniers travaux, Zhao et al. (Zhao, 2024) ont démontré que le traitement par la palmatine (PAL) améliore la fonction motrice et réduit la perte neuronale dopaminergique chez les souris induites par le MPTP. La PAL réduit la perte de neurones dopaminergiques en améliorant la mitophagie et en inhibant l'activation de l'inflammasome NLRP3, un complexe multiprotéique à l'origine de la signalisation inflammatoire. De même, Chen et al. (Chen, 2022) ont rapporté que la dexmédétomidine protège les neurones dopaminergiques chez les souris induites par le MPTP en régulant à la hausse l'expression des protéines clés de la mitophagie, renforçant ainsi la mitophagie médiée par PINK1/Parkin.
Cependant, des données provenant de modèles de souris et de drosophile, ainsi que de cultures neuronales primaires, indiquent que la perte des fonctions de PINK1 et de Parkin n'entraîne pas systématiquement l'accumulation de mitochondries dysfonctionnelles dans les cellules (Goldberg, 2003; Narendra, 2011; Ge, 2020). Ces résultats suggèrent l'existence de voies alternatives de mitophagie qui peuvent compenser les déficiences de PINK1/Parkin. L'élucidation de la manière et du moment où ces voies compensatoires sont activées permettra de mieux comprendre la vulnérabilité unique des neurones dopaminergiques dans la maladie de Parkinson par rapport à d'autres populations cellulaires.
Notre équipe se fera un plaisir de répondre à vos questions sur la mitophagie dans les maladies neurodégénératives ou de vous fournir des informations spécifiques sur les modèles de DP que nous utilisons pour les études d'efficacité thérapeutique.
En savoir plus sur nos modèles pour la maladie de Parkinson
Contenu connexe
Informations actualisées sur la mitophagie dans la maladie de Parkinson et les meilleures pratiques liées à l'évaluation des agents thérapeutiques dans les modèles animaux de maladies neurodégénératives.
Dysfonctionnement mitochondrial dans les microglies et les astrocytes
Le rôle du dysfonctionnement mitochondrial dans les microglies et les astrocytes dans les maladies neurodégénératives, notamment la maladie d'Alzheimer, la maladie de Parkinson et la SLA.
Autophagie et maladies neurodégénératives
An overview of how cellular autophagy plays a role in brain health and neurodegeneration.
Autophagie, maladie de Parkinson et neurones dopaminergiques
Une vue d'ensemble de la façon dont une autophagie déficiente peut conduire à des changements pathologiques et à la neurodégénérescence des neurones dopaminergiques dans la maladie de Parkinson.
Morphologie de la microglie dans la SLA, la maladie d'Alzheimer et la maladie de Parkinson
Une vue d'ensemble de l'analyse morphologique des microglies et des applications à la recherche sur les maladies neurodégénératives et à la découverte et au développement de médicaments.
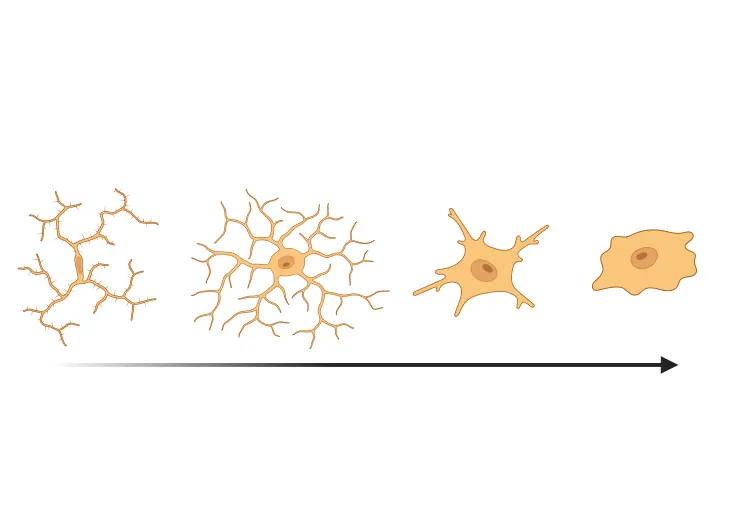
Activation microgliale dans un modèle de souris PFF à α-synucléine
Nous avons quantifié l'activation microgliale, sur la base de la morphologie, dans un modèle murin d'ensemencement et d'étalement de fibrilles préformées d'α-synucléine (PFF) de la maladie de Parkinson.
Modèles AAV d'α-synucléine pour le développement de médicaments contre la maladie de Parkinson
Vue d'ensemble des modèles de souris et de rats à α-synucléine induite par un vecteur du virus adéno-associé (AAV), à utiliser dans les études précliniques de thérapeutiques modificatrices de la maladie.
Le neurofilament à chaîne légère dans les modèles de la maladie de Parkinson
Comment les niveaux de neurofilament à chaîne légère (NfL ; NF-L) peuvent être utilisés comme biomarqueurs dans le sang et le LCR dans les modèles de souris et de rats atteints de la maladie de Parkinson.