Dysfonctionnement mitochondrial et maladie de Parkinson
Un aperçu de la façon dont le dysfonctionnement mitochondrial est associé à la neurodégénérescence dans la maladie de Parkinson.
Cette ressource décrit:
- Qu'est-ce que le dysfonctionnement mitochondrial?
- Quelle est la relation entre le dysfonctionnement mitochondrial et la dégénérescence des neurones dopaminergiques dans la maladie de Parkinson?
- Existe-t-il des approches thérapeutiques ciblant le dysfonctionnement mitochondrial dans la maladie de Parkinson?
Qu'est-ce que le dysfonctionnement mitochondrial?
Les mitochondries sont des organites à double membrane qui jouent un rôle central dans la gestion des activités métaboliques des cellules par la production d'adénosine triphosphate (ATP), la principale source d'énergie qui alimente les fonctions cellulaires (Casanova, 2023; Chen, 2023). La membrane mitochondriale interne abrite un ensemble de protéines essentielles au transport des électrons et à la synthèse de l'ATP par phosphorylation oxydative, le processus de libération de l'énergie chimique qui est convertie en ATP. La membrane mitochondriale externe (MMO), quant à elle, est perméable aux petites molécules et agit comme un tamis, permettant le passage des métabolites nécessaires à la phosphorylation oxydative. Au-delà de leur rôle d'usines énergétiques, ces organites hautement dynamiques sont également impliqués dans d'autres fonctions essentielles, telles que la régulation de l'homéostasie calcique, la signalisation épigénétique, la signalisation métabolique et la mort cellulaire programmée (Bhatti, 2017; Collier, 2023; Nyugen, 2023).
Les origines de toutes les mitochondries peuvent être retracées jusqu'à un organite ancestral commun, car les mitochondries ne peuvent pas être formées de novo (Shiota, 2015; Roger, 2017). Au contraire, elles existent au sein d'un système complexe interconnecté, où elles se divisent indépendamment du cycle cellulaire par fisson, un processus qui permet d'augmenter la production d'énergie en réponse à la demande croissante d'énergie cellulaire. Inversement, la fusion mitochondriale permet à deux petites mitochondries de former une mitochondrie plus grande, favorisant l'échange de ressources et d'autres composants entre les mitochondries pour un fonctionnement optimal. Par conséquent, le maintien d'une population mitochondriale saine nécessite l'élimination sélective des mitochondries endommagées et dysfonctionnelles du réseau mitochondrial par la mitophagie, un type d'autophagie sélective, qui marque, dégrade et recycle les mitochondries dysfonctionnelles (Ashrafi, 2013; Uoselis, 2023).
Le dysfonctionnement mitochondrial, caractérisé par une altération de la fonction mitochondriale et une incapacité à satisfaire les besoins énergétiques cellulaires, est impliqué dans la pathogenèse de plusieurs maladies neurodégénératives, dont la maladie de Parkinson (MP), la maladie d'Alzheimer (MA), la maladie de Huntington (MH) et la sclérose latérale amyotrophique (SLA) (Beal, 2000 ; Johri, 2012; Norat, 2020). Les neurones, en raison de leur forte demande énergétique et de leur dépendance à l'égard des mitochondries pour le tamponnage du calcium, sont particulièrement vulnérables à une altération de la fonction mitochondriale (Paß, 2021; Henrich, 2023).
Plusieurs facteurs contribuent au dysfonctionnement mitochondrial et l'exacerbent dans les maladies neurodégénératives, notamment:
- Une mitophagie réduite ou excessive, qui se traduit par une population mitochondriale inadéquate
- Des perturbations dans la disponibilité ou le transport des substrats nécessaires à la production d'ATP en raison de défauts dans la chaîne de transport d'électrons et la machinerie de synthèse de l'ATP
- Une production excessive d'espèces réactives de l'oxygène (ROS), qui amplifie les lésions mitochondriales et induit une mitophagie excessive
- Une régulation calcique déficiente, qui entraîne des perturbations dans la signalisation cellulaire
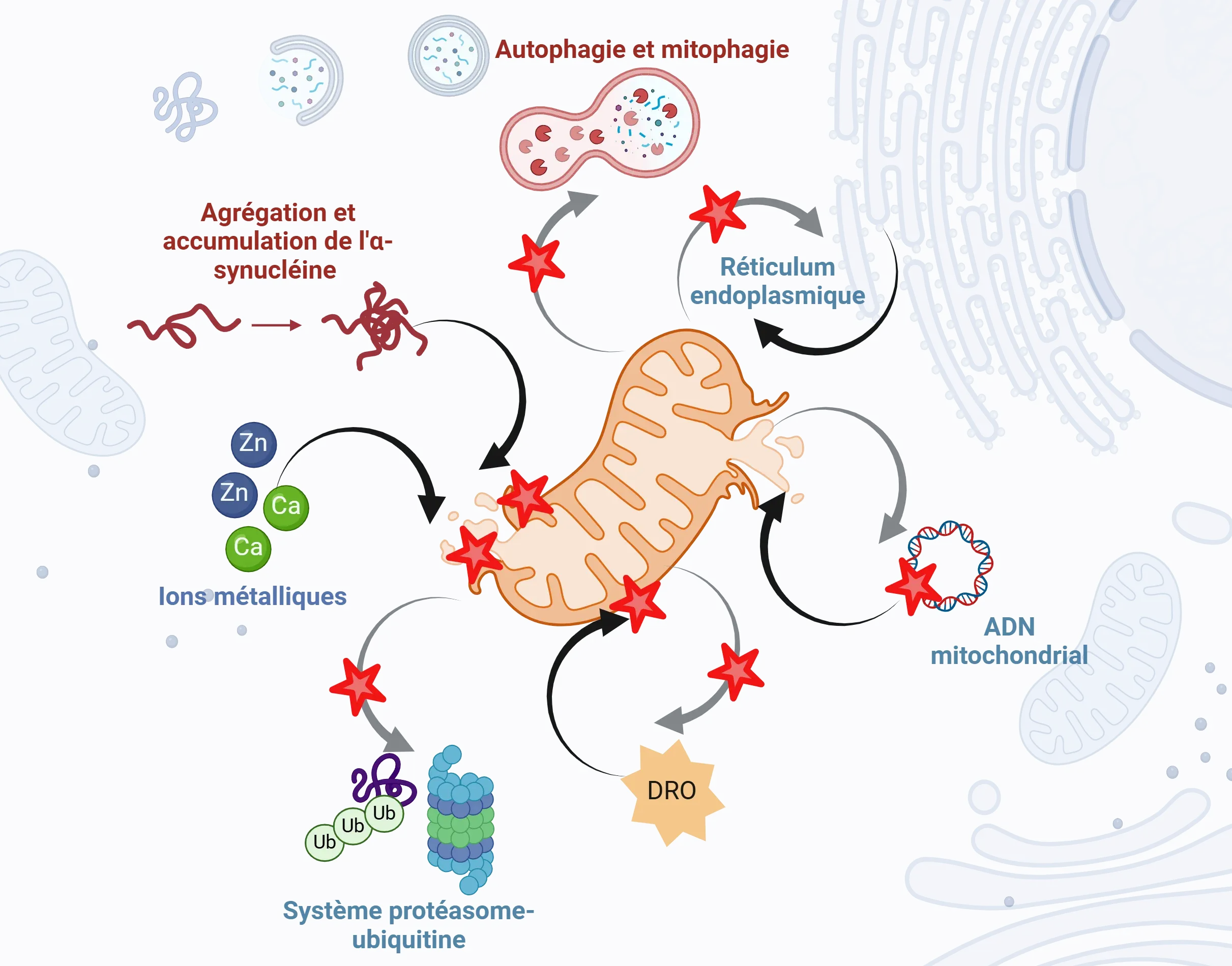
Le dysfonctionnement mitochondrial joue un rôle central, mais complexe, dans la pathogenèse de la maladie de Parkinson. Le diagramme schématique met en évidence les mécanismes communs du dysfonctionnement mitochondrial, les flèches noires décrivant les influences directes et indirectes, telles que l'agrégation des protéines, le déséquilibre Zn et Ca, les ROS et les lésions de l'ADNmt. La figure et la légende sont reproduites de Prasuhn et al. (Prasuhn, 2021) sous la licence Creative Commons Attribution.
Quelle est la relation entre le dysfonctionnement mitochondrial et la dégénérescence des neurones dopaminergiques dans la maladie de Parkinson?
La dégénérescence des neurones dopaminergiques dans la MP est étroitement liée au dysfonctionnement mitochondrial. En raison de leurs besoins énergétiques exceptionnellement élevés, ces neurones dépendent d'un réseau mitochondrial vaste et complexe qui s'étend des dendrites aux terminaisons synaptiques. La phosphorylation oxydative mitochondriale, essentielle à la production d'ATP, est altérée en cas de dysfonctionnement mitochondrial. Cependant, un apport continu d'ATP est nécessaire pour maintenir l'homéostasie ionique, stimuler l'activité synaptique et le recyclage des transmetteurs - des processus qui soutiennent la fonction et la survie des neurones dopaminergiques (Sheng, 2017; Duarte, 2023; Henrich, 2023). En outre, les mitochondries jouent également un rôle essentiel dans le tamponnage du calcium intracellulaire, qui est indispensable au bon fonctionnement des axones (Walters, 2023). Par conséquent, les perturbations des processus mitochondriaux susmentionnés ont un impact négatif sur la santé neuronale et sont des facteurs clés de la pathologie de la maladie de Parkinson.
Le dysfonctionnement mitochondrial dans la MP est encore aggravé par la pathologie de l'alpha-synucléine, une caractéristique de la MP (Geibl, 2024). Certaines espèces d'alpha-synucléine, par exemple, se lient à la protéine TOM20 de la membrane mitochondriale externe, empêchant son interaction avec son corécepteur TOM22 et altérant l'importation des protéines mitochondriales (Di Maio, 2016). La surexpression de l'alpha-synucléine induit également l'ouverture du pore de transition de perméabilité mitochondriale (mPTP), un canal protéique qui maintient l'intégrité des mitochondries (Parihar, 2008; Ludtmann, 2018). Cette ouverture entraîne une perte du potentiel de la membrane mitochondriale, une réduction de la production d'ATP, la libération d'espèces réactives de l'oxygène (ROS) nocives et la mort cellulaire.
Les mutations des gènes codant pour les protéines de contrôle de la qualité mitochondriale sont également fortement associées à la dégénérescence des neurones dopaminergiques (de Castro, 2011). Par exemple, les souris knock-out Parkin présentent une capacité respiratoire mitochondriale réduite, tandis que les souris knock-out PINK1 présentent des déficits de la capacité de tamponnage du calcium et des altérations du potentiel membranaire mitochondrial (Palacino, 2004; Akundi, 2011). Les mutations génétiques des protéines liées à la fonction lysosomale, telles que GBA1, LRRK2, VPS35, contribuent également à la perte neuronale. Des études menées sur des souris DAT-Cre ont révélé que des mutations de perte de fonction dans ces gènes perturbent la dynamique mitochondriale, entraînant une neurodégénérescence semblable à la maladie de Parkinson (Tang, 2015).
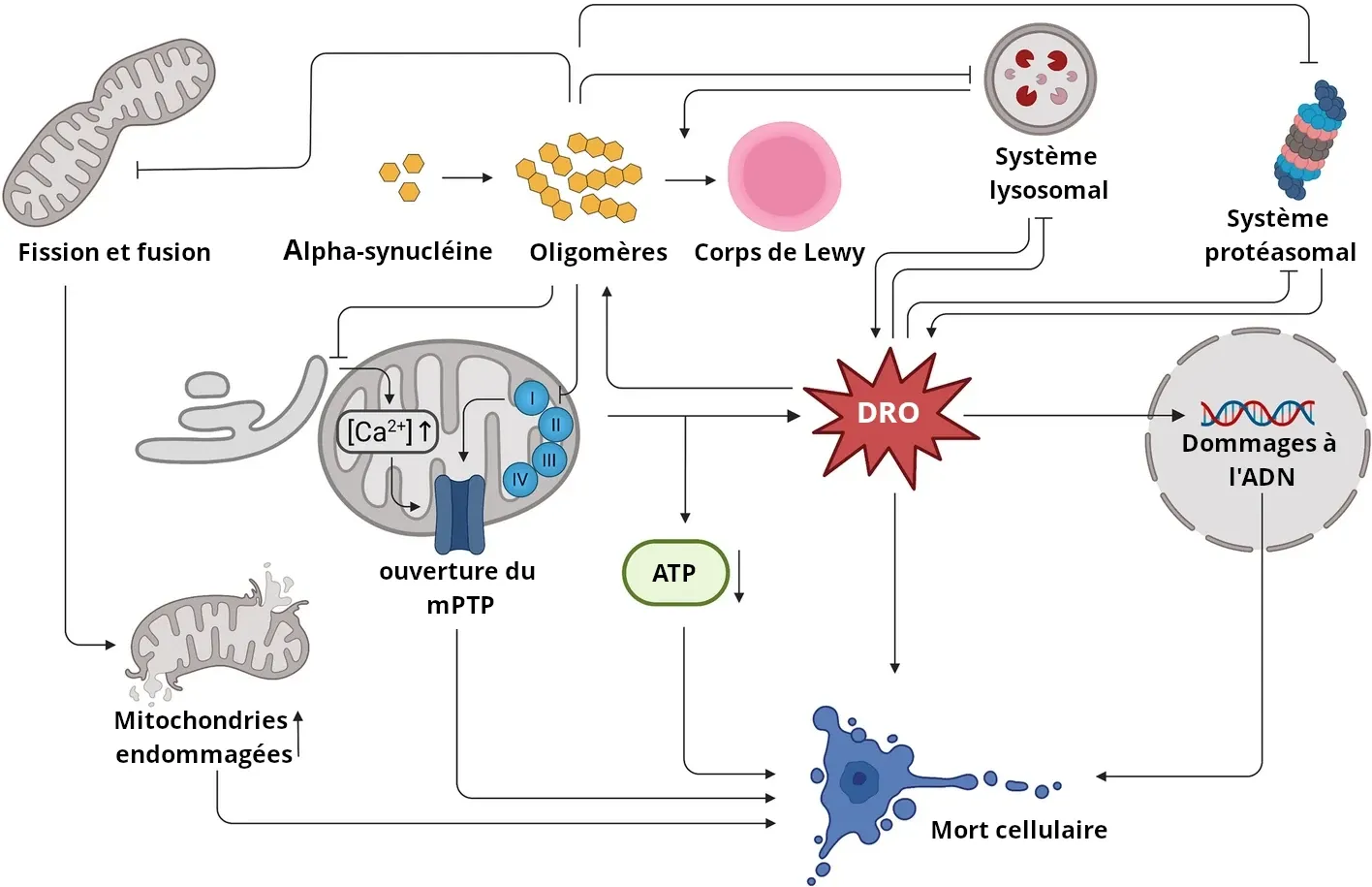
Le dysfonctionnement mitochondrial dans la MP est exacerbé par la pathologie de l'alpha-synucléine. Le diagramme décrit plusieurs mécanismes de dysfonctionnement et de mort cellulaires induits par l'alpha-synucléine. La figure est reproduite de Henrich et al. (Henrich, 2023) sous la licence Creative Commons Attribution.
Existe-t-il des approches thérapeutiques ciblant le dysfonctionnement mitochondrial dans la maladie de Parkinson?
Étant donné le lien étroit entre le dysfonctionnement mitochondrial et la neurodégénérescence, plusieurs approches thérapeutiques sont actuellement à l'étude pour atténuer les dommages mitochondriaux dans la maladie de Parkinson. Ces stratégies sont les suivantes:
Les activateurs de la mitophagie
Il s'agit de petites molécules qui activent directement les protéines critiques de contrôle de la qualité des mitochondries, PINK1 et Parkin, ou qui inhibent la fonction du contre-joueur de Parkin, la protéase spécifique de l'ubiquitine USP30. L'urolithine A, le riboside de nicotinamide, l'actinonine, la tomatidine et la spermidine sont des activateurs de la mitophagie dont le potentiel thérapeutique est actuellement à l'étude. Dans des modèles précliniques de neurodégénérescence, il a été démontré que ces composés améliorent la mitophagie en facilitant l'élimination des mitochondries défectueuses et dysfonctionnelles des cellules affectées, ce qui entraîne une amélioration des fonctions synaptiques et cognitives (Miller, 2019; Kshirsagar, 2021; Wang, 2022). En outre, des médicaments approuvés par la FDA, tels que la rapamycine et la metformine, sont également étudiés pour leur potentiel d'amélioration de la mitophagie (Sarkar, 2007). La rapamycine agit en inhibant la voie de signalisation mTOR, qui est un régulateur négatif de l'autophagie. La metformine, quant à elle, active la voie de signalisation AMPK, qui déclenche une mitophagie dépendante de PINK1/Parkin (Bharath, 2020).
Inhibiteurs du pore de transition de la perméabilité mitochondriale (mPTP)
L'ouverture du pore de transition de la perméabilité mitochondriale est un facteur clé de la mort cellulaire dans les maladies neurodégénératives (Bauer, 2020). La cyclophiline D (CypD) est la molécule la plus cruciale pour la formation du pore de transition de la perméabilité murale et agit comme une protéine régulatrice de la mort cellulaire. Les inhibiteurs de petites molécules ciblant l'activité de la CypD sont apparus comme une stratégie thérapeutique attrayante pour prévenir la transition de perméabilité, atténuer le dysfonctionnement mitochondrial et ralentir la perte neuronale dans la MP. La ciclosporine A (CsA) est l'un des inhibiteurs de CypD dont le potentiel thérapeutique est actuellement étudié dans des modèles précliniques de neurodégénérescence (Samanta, 2024).
Antioxydants ciblant les mitochondries
Le dysfonctionnement des mitochondries est un facteur clé des ROS. Leur surproduction peut entraîner un stress oxydatif accru, des dommages à l'ADN mitochondrial, des perturbations du métabolisme énergétique neuronal et une réduction de la viabilité cellulaire. Ces facteurs jouent un rôle critique dans la progression des maladies neurodégénératives (Andersen, 2004; Szeto, 2006; Dash, 2024).
Afin de lutter contre les effets néfastes des ROS, les scientifiques développent des antioxydants à visée mitochondriale qui s'accumulent sélectivement dans les mitochondries dysfonctionnelles, où ils peuvent neutraliser les ROS à la source, prévenir les dommages oxydatifs mitochondriaux et rétablir l'approvisionnement énergétique des neurones (Jin, 2014; Apostolova, 2015). Certains antioxydants ciblant les mitochondries font actuellement l'objet de recherches sur leur activité et leur potentiel thérapeutique:
- Mitoquinone Mesylate (MitoQ)
Un dérivé de l'ubiquinone conjugué au triphénylphosphonium. Le MitoQ pénètre dans la matrice mitochondriale, piège les radicaux libres et réduit leur concentration (Duarte, 2023). - Vitamine E ciblant les mitochondries (MitoVitE)
La fraction chromanol de la vitamine E est liée à un cation triphénylphosphonium. Elle pénètre à travers les bicouches lipidiques et s'accumule dans les mitochondries (Smith, 1999). - Apocynine ciblant les mitochondries (MitoApocynine)
Dérivé de l'apocynine conjugué au triphénylphosphonium. Il inhibe la production de radicaux superoxydes, qui sont une cause majeure de stress oxydatif cellulaire (Ghosh, 2016).
Thérapie génique
Les mutations de perte de fonction des gènes codant pour les protéines de contrôle de la qualité des mitochondries PINK1 et Parkin peuvent entraîner une altération de la mitophagie et une élimination insuffisante des mitochondries dysfonctionnelles. Par conséquent, les thérapies d'augmentation génique qui stimulent la voie PINK1/Parkin ont le potentiel d'améliorer la mitophagie et d'atténuer le dysfonctionnement mitochondrial (Quinn, 2020).
Notre équipe se fera un plaisir de répondre à vos questions sur le dysfonctionnement mitochondrial dans les maladies neurodégénératives ou de vous fournir des informations spécifiques sur les modèles de la maladie de Parkinson que nous utilisons pour les études d'efficacité thérapeutique.
Découvrez nos modèles de maladies neurodégénératives
Contenu connexe
Des informations actualisées sur le dysfonctionnement mitochondrial et la maladie de Parkinson dans les maladies neurodégénératives et les meilleures pratiques liées à l'évaluation des agents thérapeutiques dans les modèles animaux de maladies neurodégénératives.
Dysfonctionnement mitochondrial dans les microglies et les astrocytes
Le rôle du dysfonctionnement mitochondrial dans les microglies et les astrocytes dans les maladies neurodégénératives, notamment la maladie d'Alzheimer, la maladie de Parkinson et la SLA.
Mitophagie et maladie de Parkinson
Une vue d'ensemble de la façon dont une mitophagie déficiente peut conduire à la neurodégénérescence dans la maladie de Parkinson.
Autophagie, maladie de Parkinson et neurones dopaminergiques
Une vue d'ensemble de la façon dont une autophagie déficiente peut conduire à des changements pathologiques et à la neurodégénérescence des neurones dopaminergiques dans la maladie de Parkinson.
Autophagie et maladies neurodégénératives
An overview of how cellular autophagy plays a role in brain health and neurodegeneration.