Dysfonctionnement lysosomal dans les microglies et les astrocytes
Un aperçu du dysfonctionnement lysosomal dans la microglie et les astrocytes, et de son rôle dans les maladies neurodégénératives.
Cette ressource décrit:
- Quelquel est le rôle des lysosomes dans la microglie et les astrocytes?
- Comment le dysfonctionnement lysosomal contribue-t-il à la maladie d'Alzheimer, à la maladie de Parkinson et à la sclérose latérale amyotrophique?
- Comment le dysfonctionnement lysosomal est-il ciblé pour traiter les maladies neurodégénératives?
Quel est le rôle des lysosomes dans les astrocytes et la microglie?
Le lysosome est un organite cytoplasmique principalement connu pour son rôle dans la dégradation de divers débris cellulaires (Perera, 2016). La biogenèse des lysosomes est principalement régulée par le facteur de transcription EB (TFEB), qui contrôle l'expression des gènes responsables des protéines et des enzymes lysosomales. Le lysosome maintient une lumière acide via la pompe à protons vacuolaire-ATPase (v-ATPase), qui est contenue par une membrane spécialisée constituée de protéines membranaires associées aux lysosomes (LAMP). De nombreuses enzymes hydrolytiques qui assurent ses fonctions cataboliques sont logées dans le lysosome.
Les lysosomes sont importants pour la dégradation des matériaux extracellulaires et intracellulaires (Perera, 2016). L'absorption des matériaux extracellulaires se fait par endocytose, au cours de laquelle les déchets sont empaquetés dans un endosome qui fusionne ensuite avec le lysosome pour être dégradés. En revanche, les matériaux intracellulaires sont dégradés et recyclés par autophagie, au cours de laquelle les organites anciens ou endommagés sont empaquetés dans des autophagosomes qui fusionnent avec le lysosome pour être recyclés ou dégradés. Ainsi, les lysosomes servent de point terminal commun où les macromolécules sont décomposées en leurs constituants pour être éliminées ou recyclées.
Les astrocytes et la microglie sont essentiels pour soutenir et réguler la signalisation neuronale. Les astrocytes jouent un rôle clé dans le recyclage des neurotransmetteurs en excès pour réguler la signalisation neuronale, ainsi que dans le maintien de la barrière hémato-encéphalique, entre autres fonctions. La microglie est la principale cellule immunitaire du système nerveux central (SNC). Les cellules phagocytaires de la microglie sont importantes pour l'élimination des agents pathogènes et des débris cellulaires, y compris les agrégats de protéines présents dans les maladies neurodégénératives (Kreher, 2021). En tant que principal critère d'évaluation de la dégradation des débris cellulaires, les lysosomes sont essentiels au fonctionnement optimal des astrocytes et de la microglie (Kreher, 2021).
Pour plus d'informations sur la manière dont l'autophagie microgliale altérée conduit à des maladies neurodégénératives, veuillez consulter: Autophagie microgliale altérée dans les maladies neurodégénératives
Voir également: Autophagie, maladie de Parkinson et neurones dopaminergiques
Comment le dysfonctionnement lysosomal contribue-t-il à la MA, à la MP et à la SLA?
Le dysfonctionnement lysosomal peut se manifester de multiples façons. Les dysfonctionnements de l'acidification lysosomale, de l'activité des hydrolases lysosomales, de la perméabilité de la membrane lysosomale et de la fusion perturbée des autophagosomes ou des endosomes avec le lysosome sont couramment observés dans les maladies neurodégénératives (Kreher, 2021; Quick, 2023). Ces dysfonctionnements lysosomaux entraînent une dégradation inefficace des débris cellulaires qui peut conduire à une pathogenèse plus poussée.
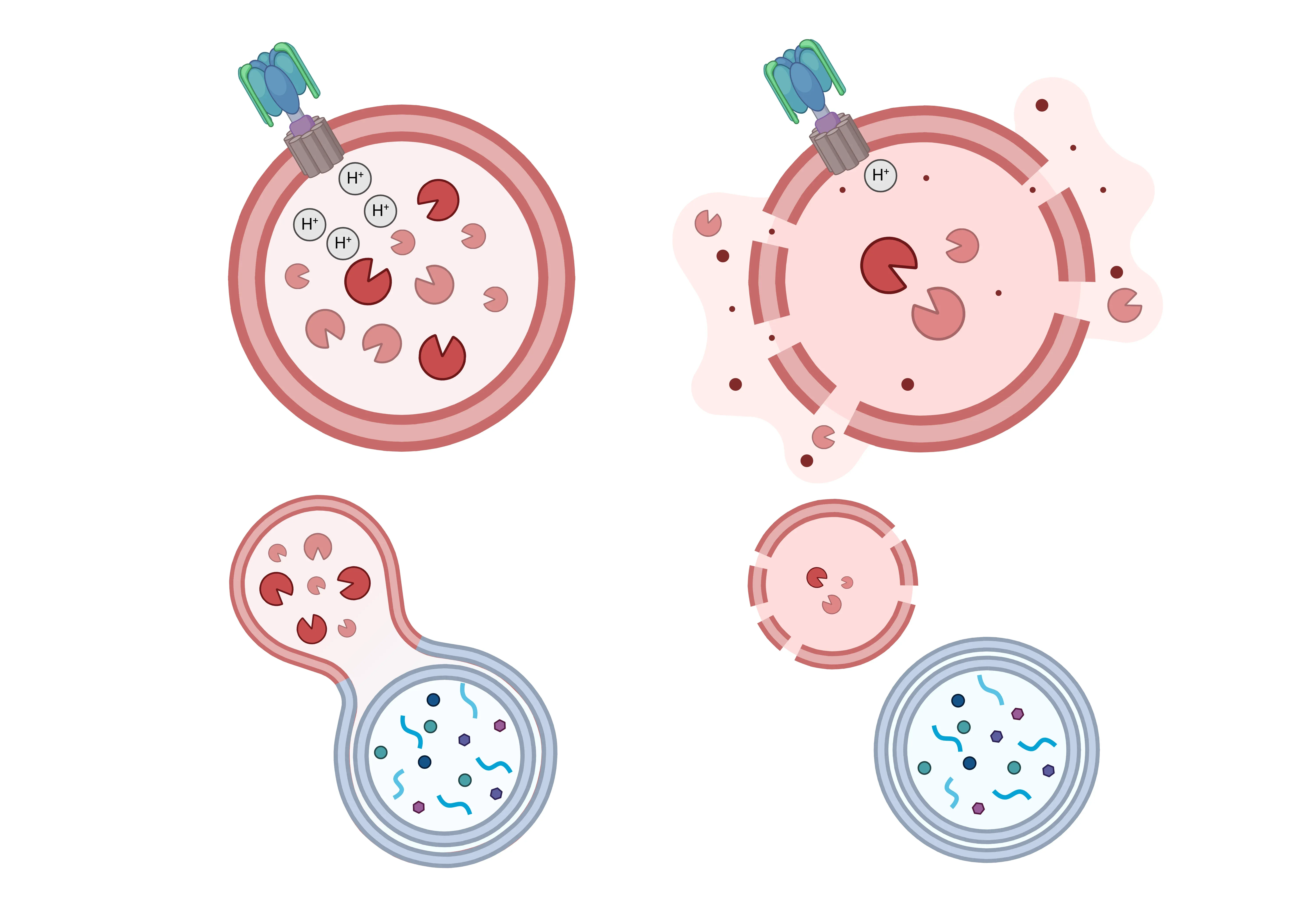
La fonction lysosomale normale(à gauche) est compromise dans de nombreuses maladies neurodégénératives et est associée à différentes manifestations de dysfonctionnement lysosomal(à droite). L'acidification lysosomale peut être affectée par des déficits de la fonction v-ATPase conduisant à une augmentation du pH dans la lumière du lysosome. De plus, la membrane lysosomale devient plus perméable, entraînant la libération cytosolique des hydrolases lysosomales. La disponibilité des enzymes lysosomales est également réduite, ce qui entraîne une diminution de la capacité de dégradation. Enfin, la fusion des lysosomes avec les endosomes et les autophagosomes peut être compromise, ce qui entraîne l'accumulation de matériaux non dégradés.
 Cliquez pour copier le lien
Cliquez pour copier le lien
La maladie d'Alzheimer (MA)
La maladie d'Alzheimer est une maladie neurodégénérative caractérisée par l'accumulation d'agrégats amyloïdes (Aβ) et tau. Les lysosomes des astrocytes et de la microglie jouent un rôle important dans l'internalisation et la dégradation de ces agrégats de protéines toxiques (Kreher, 2021). Cependant, la phagocytose et la dégradation des Aβ semblent être plus importantes dans la microglie (Prakash, 2021). Les souris dont la microglie est déficiente en autophagie présentent une réduction des enzymes lysosomiales disponibles et une clairance altérée des Aβ (Choi, 2023). En outre, il existe une perte d'enzymes lysosomiales dans la MA, ce qui entraîne une réduction de la capacité de dégradation des agrégats de protéines (Kim, 2025). Une acidification lysosomale correcte est impérative pour que le lysosome remplisse sa fonction catabolique et est altérée dans la MA. La neutralisation génétique de l'antiporteur de chlorure et d'ions hydrogène ClC-7 entraîne une acidification incorrecte des lysosomes microgliaux, ce qui entraîne une dégradation altérée des agrégats de protéines Aβ (Majumdar, 2011). En outre, les mutations de la préséniline 1 (PS1), qui ont été fortement liées au développement de la MA, entraînent des altérations de l'assemblage de la v-ATPase lysosomale, ce qui entraîne une acidification lysosomale inappropriée, ainsi qu'une dégradation inefficace de la Aβ par la microglie (Ledo, 2021). En outre, les mutations PS1 ont également été associées à une réduction des niveaux d'ARNm TFEB dans la microglie, entraînant une réduction de la biogenèse lysosomale et une altération de l'autophagie (Ledo, 2021).
La MA et l'accumulation d'agrégats de protéines peuvent également exacerber le dysfonctionnement lysosomal dans la glie. Par exemple, la présence d'agrégats d'Aβ affecte le trafic de TFEB, ce qui entraîne une acidification lysosomale inappropriée et une clairance réduite des agrégats de protéines (Guo, 2017). Une exposition prolongée aux agrégats Aβ entraîne une inhibition supplémentaire des capacités autophagiques et lysosomales des microglies en augmentant la perméabilité de la membrane lysosomale (Pomilio, 2020). Ainsi, un mécanisme de rétroaction de la dysfonction lysosomale et de l'accumulation d'agrégats de protéines neurotoxiques peut entraîner une neurodégénérescence supplémentaire dans la MA.
Maladie de Parkinson (MP)
La maladie de Parkinson est la deuxième maladie neurodégénérative la plus répandue après la maladie d'Alzheimer et se caractérise par une altération marquée de la fonction motrice ainsi que par des caractéristiques non motrices. La principale caractéristique neuropathologique de la maladie de Parkinson est la mort des neurones dopaminergiques dans la substance noire. La maladie de Parkinson se caractérise également par la présence d'agrégats de protéines α-synucléine (α-Syn), qui est le principal composant des corps de Lewy. Les agrégats de protéines dans la MP se trouvent principalement dans les neurones, mais également dans les astrocytes et la microglie (Tremblay, 2019). Les astrocytes et la microglie peuvent jouer un rôle important dans l'internalisation de la α-synucléine sécrétée par les neurones pour être dégradée de manière lysosomale (Lee, 2010; Choi, 2020). Bien que les microglies soient les principaux piégeurs responsables de l'absorption de la α-syn (Filippini, 2019), une absorption excessive de la α-syn par les astrocytes peut submerger la voie lysosomale, entraînant des dépôts de α-syn spécifiquement dans les astrocytes (Tremblay, 2019).
La leucine-rich repeat kinase 2 (LRRK2) et la glucocérébrosidase (GBA) sont deux des gènes les plus fréquemment associés à la MP (Pang, 2022), et tous deux jouent un rôle important dans les fonctions lysosomales des astrocytes et des microglies (Tremblay, 2019). Dans les astrocytes, LRRK2 est localisé avec les protéines membranaires associées aux lysosomes (LAMP), qui sont importantes pour faciliter la fusion des lysosomes avec les endosomes et les autophagosomes pour la dégradation (Henry, 2015; Tremblay, 2019). Les mutations de LRRK2, qui ont été liées à la MP, présentent des astrocytes avec des lysosomes hypertrophiés et une capacité de dégradation réduite (Henry, 2015). Il a été démontré que LRRK2 régule négativement la dégradation lysosomale dans les astrocytes et la microglie, potentiellement par ses effets sur TFEB (Henry, 2015; Yadavalli, 2023). Une activité LRRK2 plus élevée diminue le nombre de lysosomes dans les astrocytes et réduit l'expression des hydrolases lysosomales dans la microglie (Henry, 2015; Yadavalli, 2023). Le gène GBA code pour l'enzyme lysosomale glucocérébrosidase (GCase), qui a également été communément associée à la MP. Le GBA est impliqué dans la mobilisation des astrocytes et de la microglie en réponse à la neurodégénérescence (Tremblay, 2019). En outre, la suppression du GBA réduit l'activité de la GCase et entraîne un dysfonctionnement lysosomal, ce qui peut augmenter l'accumulation d'agrégats de α-Syn (Schöndorf, 2014). Par conséquent, les deux gènes les plus associés à la MP semblent avoir un impact sur la fonction lysosomale au sein des astrocytes et de la microglie.
Sclérose latérale amyotrophique (SLA)
La SLA est une maladie mortelle caractérisée par la dégénérescence des motoneurones dans le SNC. Il a été découvert qu'une expansion de répétitions d'hexanucléotides dans la région non codante du gène du cadre de lecture ouvert 72 du chromosome 9 (C9orf72) est l'une des principales causes génétiques sous-jacentes de la maladie (Root, 2021). Il est intéressant de noter que les mutations du gène C9orf72 se sont également avérées jouer un rôle important dans la fonction lysosomale. Les protéines C9orf72 sont localisées dans le lysosome (Amick, 2016; Laflamme, 2019) et sont fortement exprimées dans la microglie (O'Rourke, 2016). Les mutations C9orf72 peuvent perturber l'acidification lysosomale, entraînant une dégradation altérée (Shao, 2019). De plus, les souris C9orf72 knock-out présentent un trafic lysosomal altéré pour la dégradation spécifiquement dans la microglie (O'Rourke, 2016). Le dysfonctionnement de la voie de dégradation lysosomale induit par les mutations C9orf72 peut entraîner l'accumulation d'agrégats protéiques qui contribuent à la neurodégénérescence dans la SLA (Root, 2021).
La protéine de liaison à l'ADN de réponse transactive 43 kDa (TDP-43) est une protéine codée par le gène TARDBP qui a également été associée à la SLA (Root, 2021). La TDP-43 est importante pour le fonctionnement normal des lysosomes et de la voie de dégradation lysosomale. Par exemple, la TDP-43 intervient dans l'expression de la TFEB, responsable de la régulation de la fonction et de la biogenèse lysosomales (Root, 2021). De plus, les mutations de la TDP-43 sont liées à un dysfonctionnement lysosomal (Kreiter, 2018). Il est intéressant de noter qu'il a été démontré que la perte de TDP-43 dans la microglie améliore la biogenèse lysosomale, favorisant la phagocytose microgliale et l'élimination des agrégats de protéines (Paolicelli, 2017). Les agrégats de TDP-43 peuvent également s'accumuler dans les neurones et la glie dans de nombreuses maladies neurodégénératives, dont la SLA (Root, 2021). Un excès de TDP-43 peut perturber les capacités de dégradation du lysosome, ce qui peut déclencher une autophagie aberrante et réduire la capacité à éliminer les agrégats de TDP-43 (Leibiger, 2018). Cet effet est très néfaste car l'élimination des agrégats de TDP-43 dans la SLA devient dépendante de la voie autophagie-lysosome, car les autres systèmes de dégradation sont submergés (Root, 2021).
Comment la fonction lysosomale est-elle ciblée pour traiter les maladies neurodégénératives?
Il existe des preuves solides établissant un lien entre les maladies neurodégénératives et le dysfonctionnement lysosomal. Par conséquent, les stratégies thérapeutiques qui améliorent ou restaurent la fonction lysosomale peuvent être utilisées pour réduire les symptômes pathologiques de la neurodégénérescence. Les stratégies mises en œuvre visent souvent à augmenter la clairance des agrégats de protéines en restaurant ou en améliorant la fonction lysosomale. Par exemple, un essai clinique de phase 2 contrôlé par placebo (NCT02914366) est actuellement en cours pour tester l'ambroxol, qui augmente l'activité de l'enzyme lysosomale GCase et qui s'est avéré réduire les agrégats de α-Syn chez la souris (Migdalska-Richards, 2016). Une autre piste explorée dans les essais cliniques est l'utilisation d'inhibiteurs de LRRK2 pour corriger le dysfonctionnement lysosomal dans le SNC (NCT04557800 et NCT04056689). Ces inhibiteurs de LRRK2 se sont révélés généralement sûrs et bien tolérés tant chez les témoins sains que chez les patients atteints de la maladie de Parkinson, mais leur efficacité reste à démontrer (Jennings, 2023).
Une cible courante pour le traitement des maladies neurodégénératives par le lysosome est le TFEB, qui est fortement impliqué dans la biogenèse et la fonction lysosomales (Kim, 2025). Le TFEB peut être activé par l'inhibition de la voie de la cible de la rapamycine chez les mammifères (mTOR), ce qui est réalisé par des molécules telles que la rapamycine (Kim, 2025). D'autres molécules peuvent également augmenter le TFEB par une voie indépendante de mTOR et il a été démontré qu'elles augmentent la dégradation des agrégats de protéines Aβ (Portbury, 2017). En outre, l'activation du TFEB peut être obtenue par l'inhibition d'autres processus en amont. Par exemple, l'inhibition sélective de la lipide kinase PIKfyve avec l'AIT-101 active le TFEB et facilite la clairance lysosomale des agrégats de TDP-43 et réduit la neuroinflammation et les déficits fonctionnels dans un modèle murin de SLA (Young, 2023). La poursuite du développement de molécules qui activent le TFEB et facilitent la clairance lysosomale, en particulier dans la glie, présente un grand potentiel pour le traitement des maladies neurodégénératives. L'augmentation de l'expression du TFEB et d'autres protéines impliquées dans l'autophagie améliore l'activation microgliale pour lutter contre la neuroinflammation et la clairance des agrégats de α-Syn (Chen, 2021). En outre, une plus grande expression de TFEB améliore également la biogenèse lysosomale et l'absorption par les astrocytes des agrégats d'Aβ et de Tau (Xiao, 2014; Martini-Stoica, 2018). La restauration de la fonction lysosomale dans la glie pourrait fournir de nouvelles approches dans le traitement des maladies neurodégénératives.
Notre équipe se fera un plaisir de répondre à toutes vos questions sur le dysfonctionnement lysosomal dans la microglie et les astrocytesdans dans les maladies neurodégénératives ou de vous fournir des informations spécifiques sur les modèles de MA, SLA et MP que nous utilisons pour les études d'efficacité thérapeutique.
En savoir plus sur nos modèles de maladies neurodégénératives
Contenu connexe
Informations actualisées sur le dysonctionnement lysosomal dans les microglies et les astrocytes et les meilleures pratiques liées à l'évaluation des agents thérapeutiques dans les modèles animaux de maladies neurodégénératives.
Dysfonctionnement mitochondrial dans les microglies et les astrocytes
Le rôle du dysfonctionnement mitochondrial dans les microglies et les astrocytes dans les maladies neurodégénératives, notamment la maladie d'Alzheimer, la maladie de Parkinson et la SLA.
Autophagie et facteur de transcription EB (TFEB)
Aperçu du facteur de transcription EB (TFEB) et de son rôle dans l'autophagie et les maladies neurodégénératives.
Autophagie et maladies neurodégénératives
An overview of how cellular autophagy plays a role in brain health and neurodegeneration.
Autophagie, maladie de Parkinson et neurones dopaminergiques
Une vue d'ensemble de la façon dont une autophagie déficiente peut conduire à des changements pathologiques et à la neurodégénérescence des neurones dopaminergiques dans la maladie de Parkinson.