NLRP3インフラマソームと神経変性疾患
NLRP3インフラマソームの概要と、アルツハイマー病、パーキンソン病、ALSなどの神経変性疾患におけるその役割について。
NLRP3インフラマソームとは何ですか?
NOD様受容体ファミリーピリンドメイン含有タンパク質3(NLRP3)インフラマソームは、最も広く研究されているインフラマソームであり、自然免疫系と炎症性シグナル伝達の制御に重要な役割を果たす多タンパク質複合体です。また、さまざまな免疫および炎症関連疾患の発症にも関与しています。その典型的な活性化経路は、病原体関連分子パターン(PAMPs)または損傷または細胞ストレスに応答して放出される宿主由来損傷関連分子パターン(DAMPs)が、パターン認識受容体(PRR)によって検出されると引き起こされます。NOD様受容体(NLR)、トールライク受容体(TLR)、およびメラノーマ2様受容体(ALR)は、PAMPsとDAMPsを認識し、その後のNLRP3インフラマソームの活性化に備えて細胞を準備させる、PRRの異なるタイプです。
NLRP3インフラマソームは主に3つの構成要素から成り、(1) センサーとしてのNLRP3、(2) アダプターとしてのアポトーシス関連スペック様タンパク質(C-terminal caspase recruitment domain (ASC)) 、(3) エフェクターとしてのプロカスパーゼ-1です。インフラマソームの形成プロセスは、PAMPsまたはDAMPSの検出から始まります。 ASCは次にプロカスパーゼ-1と結合し、それを活性型カスパーゼ-1に変換します。 活性型カスパーゼ-1は、プロインターロイキン(IL)-1βおよびプロIL-18を活性型であるIL-1βおよびIL-18に切断します。また、カスパーゼ-1はガスダーミンD(GSDMD)を活性化し、GSDMDは膜孔を形成して炎症性サイトカインの放出を可能にし、さらに、ピロトーシスの引き金となります。
NLRP3インフラマソームは、免疫および炎症関連疾患に加え、アルツハイマー病(AD)、パーキンソン病(PD)、筋萎縮性側索硬化症(ALS)などの神経変性疾患の病態にも関与していることが示唆されています(Holbrook, 2021;Singh, 2023)。ミクログリアとアストロサイトは、中枢神経系において当初は保護的な免疫応答を提供しますが、慢性的に活性化すると神経炎症の拡大につながり、結果としてこれらの神経変性疾患の進行を助長します(Wang, 2024)。そのため、NLRP3インフラマソームを標的とした治療法は、慢性的な炎症を軽減し、疾患の症状を緩和する可能性があるとして注目されています。
最近の研究では、NLRP3の活性化を調節する小分子を探索し、慢性炎症の抑制と疾患の予後の改善に有望な結果が示されています(Blevins, 2022)。中枢神経系疾患を対象としたNLRP3インフラマソームを標的とした効果的な治療法の開発には、血液脳関門(BBB)の回避など、依然として課題はありますが、医薬品開発と標的療法の継続的な進歩が、効果的な治療法の実現への道筋をつけるでしょう。
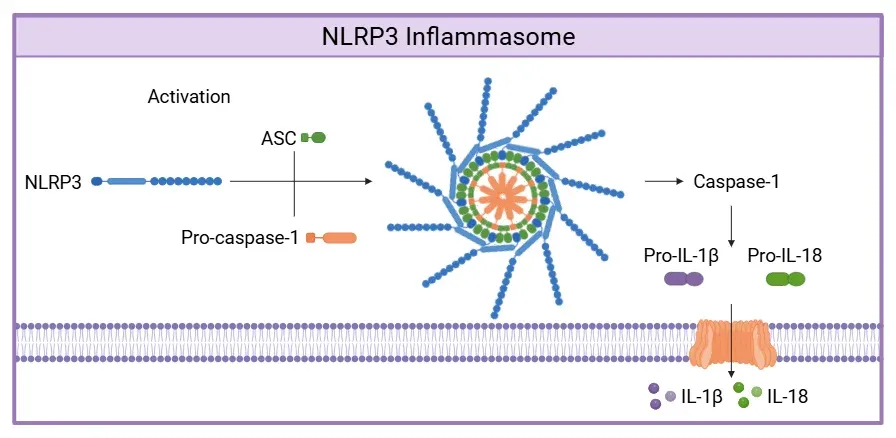
NLRP3インフラマソームは、NLRP3がPAMPsまたはDAMPsを検出すると(図示せず)、正規の経路を介して活性化されます。この認識によりASCとプロカスパーゼ-1がリクルートされ、NLRP3インフラマソーム複合体の形成につながります。形成されると、活性型カスパーゼ-1はプロIL-1βおよびプロIL-18を活性型であるIL-1βおよびIL-18、ならびにGSDMD(図示せず)に切断します。GSDMDの切断により、炎症性サイトカインであるIL-1βおよびIL-18の放出を可能にする膜孔が形成されます。図とキャプションは、Wang et al. (Wang, 2024) の著作権表示ライセンスに基づき、改変しています。
NLRP3インフラマソームは、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症においてどのような役割を果たしているのでしょうか?
アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症などの神経変性疾患は、ますます増加傾向にあります。これらの疾患は、中枢神経系のさまざまな部位における異常なタンパク質の蓄積の進行により特徴づけられ、神経細胞の損失につながります。主な関連タンパク質および病理には、アミロイドβ(Aβ)プラーク、過剰リン酸化タウ神経原繊維変化、α-シヌクレイン(α-syn)凝集体、TAR DNA結合タンパク質-43(TDP-43)およびスーパーオキシドジスムターゼ1(SOD1)封入体などがあります。
このタンパク質の蓄積がもたらす重大な結果として、NLRP3インフラマソームの活性化が挙げられます。これは、炎症性サイトカインであるIL-1βおよびIL-18の放出を誘発し、神経炎症の一因となります。 疾患の進行、炎症の増幅、神経細胞の損傷においてインフラマソームが中心的な役割を果たしていることを示す証拠が増えています。 インフラマソームの関与に関する理解が深まるにつれ、これらの疾患におけるインフラマソームの活性化を調査することがますます重要になっています。
アルツハイマー病(AD)
アルツハイマー病(AD)は、認知機能の低下、記憶障害、行動や気分に変化が現れる進行性の神経変性疾患です。 βアミロイド斑と過剰リン酸化タウ神経原繊維の蓄積が、ADの病態の中心となっています。 しかし、神経炎症もまた、病気の進行に重要な役割を果たしています。 ADでは、βアミロイドとタウがDAMPsとして作用し、特にミクログリアにおいてNLRP3インフラマソームを活性化します。この活性化は、IL-1βなどの炎症性サイトカインの放出につながり、神経炎症を悪化させ、神経変性を加速させます(Wang, 2024)。したがって、AD患者の末梢免疫細胞および死後脳組織では、IL-1βおよびIL-18のレベルが上昇し、インフラマソーム構成因子であるNLRP3、ASC、およびカスパーゼ-1の発現も増加しており、ASCの発現はAβおよびタウのレベルと相関しています(Heneka, 2013;Saresella, 2016;Vontell, 2023)。
同様に、APP/PS1 AD マウスモデルでは、カスパーゼ-1 の処理の増加が観察されています(Heneka, 2013)。これらのマウスを NLRP3 またはカスパーゼ-1 欠損モデルと交配すると、記憶が維持されることが示され、NLRP3 およびカスパーゼ-1 が AD における認知機能障害に関連する炎症を媒介することが示唆されています(Heneka, 2013)。さらに、これらのAPP/PS1/NLRP3-/-またはAPP/PS1/Casp-1-/-マウスでは、Aβの貪食が増加しており、NLRP3インフラマソームの活性化がAβの貪食を減少させることを示唆しています(Heneka, 2013)。これらの知見は、AβがNLRP3インフラマソームを活性化させ、炎症反応を促進することでアルツハイマー病の進行を早め、認知障害の一因となり、Aβの除去を妨げているという仮説を裏付けるものです。
Aβ病理に加えて、NLRP3インフラマソームの活性化はタウ病理も促進します。Tau22マウスでは、カスパーゼ-1、ASC、およびIL-1βのレベル上昇が検出されています(Ising, 2019)。NLRP3インフラマソームの活性化がADの認知機能に影響を与えるという役割と一致して、ASCまたはNLRP3欠損と交配したTau22マウスでは、タウの過剰リン酸化および凝集のレベルが低下し、記憶も維持されています(Ising, 2019)。APP/PS1 脳ホモジネートを Tau22 マウスに注入すると、タウの過剰リン酸化が誘発されますが、この効果は Tau22/ASC-/- または Tau22/NLRP3-/-マウスでは認められず、NLRP3 の活性が Aβ-タウカスケードの重要な構成要素であることを示唆しています(Ising, 2019)。Aβおよびタウ病理の両方において重要な役割を果たしていることから、NLRP3インフラマソームを標的とすることは、治療効果をもたらし、アルツハイマー病やその他のタウオパチーの進行を遅らせる可能性がある有望な戦略です(Heneka, 2013;Ising, 2019)。
パーキンソン病(PD)
アルツハイマー病に次いで2番目に多い神経変性疾患であるパーキンソン病は、主に筋肉の硬直、動作緩慢、安静時振戦などの運動症状と、うつ病などの気分障害や行動障害などの非運動症状を伴うことが特徴です。PDは、黒質緻密部(SNc)におけるドーパミン作動性(DA)ニューロンの進行性損失や、主にα-シヌクレイン凝集体で構成されるレビー小体の蓄積などの特徴的な症状によって定義されます。これらの特徴的な症状に加えて、炎症もPDの病理において重要な役割を果たしています(Li, 2021)。パーキンソン病患者の死後組織では、ASCとNLRP3のレベルが上昇していることが検出されており、この疾患におけるNLRP3インフラマソームの活性化を示唆しています(Anderson, 2021)。
MPTP誘発性のマウスパーキンソン病モデルにおいて、NLRP3またはカスパーゼ-1ノックアウトマウスを交配させると、DAニューロンの損失が減少し、運動機能が改善することが示されています(Qiao, 2017;Lee, 2019)。さらに、NLRP3欠損マウスでは、MPTPによるSNにおけるミクログリアの動員、IL-1β産生、およびカスパーゼ-1活性化が起こらないことから、ミクログリアにおけるNLRP3インフラマソームの活性化が、PDで見られる神経変性において重要な役割を果たしていることが示唆されています(Lee, 2019)。興味深いことに、選択的ドーパミンD2受容体アゴニストが、MPTP処理したパーキンソン病モデルマウスの線条体におけるNLRP3インフラマソームの活性化を阻害することが分かっています(Zhu, 2018)。さらに、NLRP3インフラマソームの活性化後の初代培養マウスアストロサイトにおいて、カスパーゼ-1とIL-1βの発現を抑制することが示されています(Zhu, 2018)。 一部の研究ではアストロサイトにおけるNLRP3インフラマソームの活性化を支持していますが(Freeman, 2017)、相反する証拠も存在しており、その関与を明らかにするにはさらなる研究が必要であることを示唆しています。
NLRP3インフラマソームの活性化は、α-シヌクレイン病理を直接調節する可能性があります。カスパーゼ-1は直接α-シヌクレインを切断することができ(Wang, 2016)、低分子阻害剤MCC950でNLRP3インフラマソームの活性化を阻害すると、PFFおよびAAV-Synマウスの両方のPD モデルにおいて、α-シヌクレインの凝集、ドーパミン変性、神経炎症、運動障害が減少します(Gordon, 2018;Grotemeyer, 2023)。これらの知見は、NLRP3インフラマソームを標的とすることが、パーキンソン病の有望な治療アプローチとなり得ることを示唆しています。
筋萎縮性側索硬化症(ALS)
ALSは、脊髄、脳幹、運動皮質における運動ニューロンの進行性変性によって特徴づけられる神経変性疾患であり、筋力低下、発声や嚥下の困難、進行性の麻痺などの症状を引き起こします。ALS患者は、C9orf72 、TARDBP 、SOD1 、FUSに変異があり、TDP-43とSOD1タンパク質が最も広く研究されています。これらのタンパク質は異常凝集体を形成し、タンパク質の除去障害と神経細胞の機能障害を引き起こします。さらに、NLRP3インフラマソームが重要な役割を果たしている可能性もあります。というのも、ALS患者の死後組織において、NLRP3、ASC、IL-18、およびカスパーゼ-1のレベル上昇が検出されているからです(Johann, 2015)。
SOD1G93AおよびTDP-43Q331KALSマウスモデルの両方において、脊髄組織ではNLRP3インフラマソーム経路遺伝子の発現が上昇しています(Deora, 2020)。SOD1G93Aマウスでは、脊髄のアストロサイトがNLRP3構成要素を発現する主要な細胞タイプであることが確認されています(Johann, 2015)。SOD1G93Aモデルにおけるカスパーゼ-1の阻害は、発症、神経学的悪化、および死亡率を遅らせることが示されており、ALSの進行におけるNLRP3インフラマソームの活性化の役割がさらに強調されています(Zhang, 2013)。しかし、他の研究では、このモデルにおいてMCC950の投与では脊髄の炎症が抑制されないことが示されており、ALSでは複数のインフラマソームが活性化される可能性があり、NLRP3の阻害だけではこの疾患の炎症を抑制するには不十分である可能性が示唆されています(Clénet, 2023)。
結論として、NLRP3インフラマソームの活性化は、ADやPDでは広く研究されている一方で、ALSにおけるその役割はまだ十分に理解されていません。NLRP3に加え、AIM2、NLRC4、NLRP1などの他のインフラマソーム複合体も、これらの神経変性疾患における神経病理学に寄与している可能性があります。神経変性疾患におけるインフラマソームの複雑な役割を解明し、潜在的な治療ターゲットを特定し、効果的な治療法を開発するためには、より多くの前臨床研究と臨床試験が必要です。
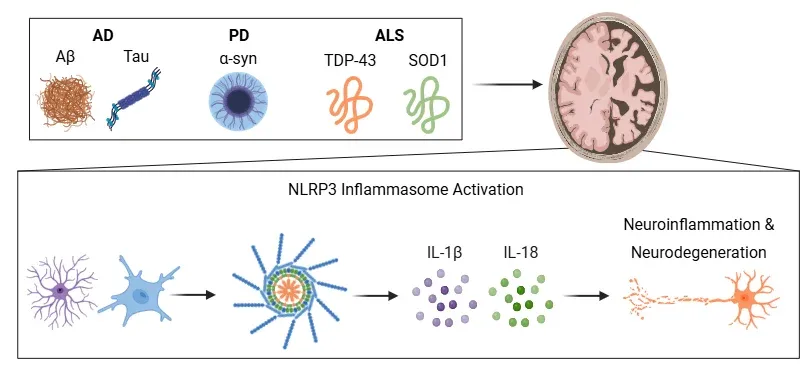
アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症などの神経変性疾患は、一般的にAβ、タウ、α-シヌクレイン、TDP-43、SOD1などの異常なフォールディングを起こしたタンパク質の異常凝集を特徴とします。これらの神経毒性タンパク質の凝集体は、ミクログリアおよび/またはアストロサイトのNLRP3インフラマソームの活性化を引き起こし、NLRP3インフラマソーム複合体の形成につながります。その結果、炎症性サイトカインであるIL-1βおよびIL-18が産生・放出され、これらの疾患における神経炎症の媒介と神経変性の促進に重要な役割を果たします。図とキャプションは、Wang et al. (Wang, 2024) の作品を改変したもので、クリエイティブ・コモンズ表示ライセンスに基づき使用しています。
神経変性疾患における NLRP3 インフラマソーム経路の治療的標的化に向けた戦略にはどのようなものがあるのでしょうか?
NLRP3インフラマソームは、神経炎症を促進することで神経変性疾患に重要な役割を果たしており、この神経炎症は疾患の進行に大きく影響しています。この発見により、神経炎症およびその有害な影響を低減させることを目的として、NLRP3インフラマソーム経路を標的とする様々な化合物の開発が進められています。
最もよく知られ、広く研究されているNLRP3阻害剤のひとつにMCC950(別名CRID3)があります。MCC950は、他のインフラマソーム複合体には影響を与えずにNLRP3インフラマソームの活性化を特異的に阻害し、IL-1βなどの炎症性サイトカインの放出を防ぎます。前臨床試験では、さまざまな神経変性疾患のマウスモデルにおいて症状を緩和する能力が示されています(Blevins, 2022)。MCC950に加え、グリブリドや OLT1177 などの他の NLRP3 阻害剤についても、その治療効果について研究が進められています。 その他のインフラマソーム阻害剤であるBAY 11-7082 やパルテノライドは、複数のインフラマソーム複合体を標的としていますが、NLRP3 に対する特異性は低くなっています。
NLRP3 カナリア型インフラマソームのシグナル伝達経路の特定の構成要素を阻害することも、別のアプローチです。選択的カスパーゼ-1 阻害剤である VX-740 および VX-765 は、IL-1βおよびIL-18の放出を阻害し、炎症を軽減します。前臨床研究では、これらの阻害剤がADマウスモデルの認知機能を改善できることが示されています(Wang, 2024)。同様に、ASC阻害剤IC100は、実験的自己免疫性脳脊髄炎(EAE)モデルにおいて抗炎症作用を示しています(Wang, 2024)。GSDMDを標的とするのも、もう一つの有望な戦略です。GSDMD ポア形成阻害剤であるジスルフィラムは、PD 細胞モデルにおける炎症の軽減に有効性を示しており、また、別の GSDM 阻害剤であるネクロスルホンアミドは、MPTP 誘発性 PD モデルにおける神経炎症の軽減と DA 神経細胞の保護作用が認められています(Wang, 2024)。
前臨床試験では有望な結果が得られているものの、神経変性疾患の治療を目的としたNLRP3阻害剤の臨床試験は依然として限られています。注目すべき研究のひとつに、ALS患者を対象に試験された低分子NLRP3阻害剤であるウスノフラスト(ZYIL1)の第2相試験(NCT05981040)があります。この試験は、IL-1βおよびIL-18の著しい阻害を示した第1相試験に続くもので、神経変性疾患の治療戦略としてのNLRP3阻害の可能性を強調するものです(Parmar, 2023)。
中枢神経系疾患に対する NLRP3 を標的とした治療法の開発における主な課題は、重大な障害となる血液脳関門を越えることです。さらに、IL-1βシグナル伝達を阻害すると感染に対する感受性が高まる可能性があるため、治療効果と安全性のバランスを取る必要があります。これらの課題は、薬物動態学的および選択性の限界を克服するために、NLRP3 の低分子阻害剤が必要であることを強調しています(Blevins, 2022)。
まとめると、NLRP3インフラマソームを標的とすることは神経変性疾患の治療に有望ですが、特異性、血液脳関門の通過、潜在的な副作用について慎重に考慮する必要があります。これらの戦略の治療可能性を判断するには、現在進行中の研究と臨床試験が極めて重要となります。
NLRP3 インフラマソームと神経変性疾患に関するご質問や、治療効果研究に使用しているAD、ALS、PDモデルに関する具体的な情報については、お気軽にお問い合わせください。
神経変性疾患モデルについてさらに詳しく知る
関連コンテンツ
NLRP3インフラムマソームと神経変性疾患に関する最新情報、および神経変性疾患モデル動物における治療薬の評価に関するベストプラクティス。
インフラマソームとは何ですか?
炎症小体(インフlammasome)の概要:その作用機序、疾患における役割、および薬物開発における標的化について。
NLRP3とは何ですか?
NLRP3およびNLRP3炎症小体活性化トリガー、疾患関連、および治療的標的戦略の概要。
インターロイキン-1ベータ(IL-1β)と神経変性疾患
アルツハイマー病(AD)、パーキンソン病(PD)、筋萎縮性側索硬化症(ALS)などの神経変性疾患におけるIL-1βの役割。
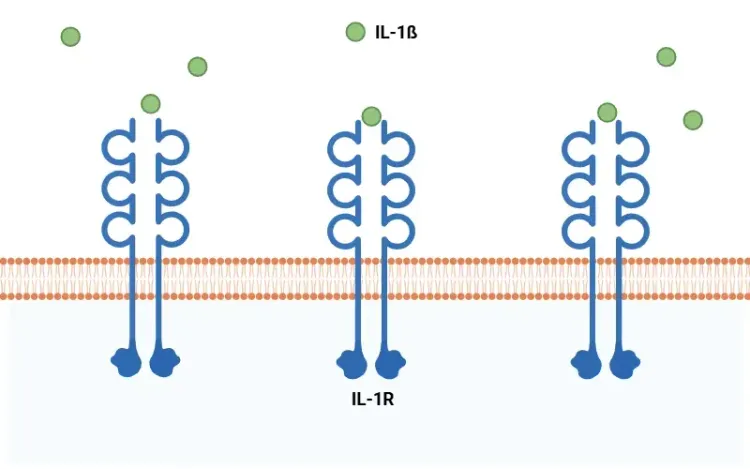
IL-1βとは何ですか?
IL-1βの概要、全身性疾患および神経疾患におけるその炎症誘発作用、ならびにIL-1β拮抗作用を基盤とした治療戦略について概説します。
ミクログリアとアストロサイトのライソゾーム機能不全
ミクログリアとアストロサイトのライソゾーム機能不全の概要と、神経変性疾患におけるその役割。